1.本发明属于医药化工领域,涉及曲拉西利新中间体及其制备以及用其制备曲拉西利关键中间体。
背景技术:
2.曲拉西利(trilaciclib,商品名:科赛拉)是先声药业与美国生物制药公司(g1 therapeutics)合作研发的一款细胞周期蛋白依赖性激酶4/6(cdk4/6)抑制剂,帮助保护骨髓中的造血干细胞和祖细胞(hspc)免受化疗诱导的骨髓抑制。2020年8月,先声药业向g1公司购买了曲拉西利在中国所有适应症的开发和商业化权利。2021年2月,曲拉西利在美国获批上市,用于既往未接受过系统性化疗的广泛期小细胞肺癌患者,在接受含铂类药物联合依托泊苷方案治疗前预防性给药,以降低化疗引起的骨髓抑制的发生率。曲拉西利的上市,成为全球首个且唯一具有全面骨髓保护功效的药物,解决化疗导致的骨髓抑制剂这一长期面临的困扰,为更多患者带来了治疗希望,其他适应症也处于临床研究中。
3.曲拉西利的化学名为:2'-((5-(4-甲基哌嗪-1-基)吡啶-2-基)氨基)-7',8'-二氢-6'h-螺[环己烷-1,9'-吡嗪并[1',2':1,5]吡咯并[2,3-d]嘧啶]-6'-酮,其关键中间体结构式为:2'-氯-7',8'-二氢-6'h-螺[环己烷-1,9'-吡嗪并[1',2':1,5]吡咯并[2,3-d]嘧啶]-6'-酮,其中它们的结构式如下:
[0004][0005]
wo2012061156a报道了曲拉西利关键中间体的合成方法,利用5-溴-2,4-二氯嘧啶为起始原料,先与1-((叔丁氧羰基)氨基甲基)环己-1-胺缩合,然后与3,3-二乙氧基丙-1-炔发生sonogashira偶联反应,接着在四丁基氟化铵(tbaf)作用下成环,再利用醋酸脱去缩醛保护基后再利用oxone氧化醛基得到7-(1-((叔丁氧羰基)氨基甲基)环己基)-2-氯-7h-吡咯并[2,3-d]嘧啶-6-甲酸,最后在dcc/dmap/三氟乙酸体系中一锅法脱boc并形成内酰胺成环得到目标产物2'-氯-7',8'-二氢-6'h-螺[环己烷-1,9'-吡嗪并[1',2':1,5]吡咯并[2,3-d]嘧啶]-6'-酮,合成路线如下所示:
[0006][0007]
该路线原料3,3-二乙氧基丙-1-炔单价较高,将其缩醛转化为羧酸需要通过酸解、氧化两步反应,进一步增加路线成本,另外oxone氧化反应在工艺放大时也具有一定安全隐患。总之,从整体上看该路线路线过长、步骤繁琐,总体收率较低,不适合放大生产。
[0008]
专利公开号cn111867592a改进了合成方法,利用的5-碘-2,4-二氯嘧啶为起始原料,先与1-((叔丁氧羰基)氨基甲基)环己-1-胺缩合,然后利用活性较高的丙炔酸甲酯代替3,3-二乙氧基丙-1-炔参与sonogashira偶联反应,接着在tbaf作用下成环,最后在三氟乙酸作用下一锅法脱boc并成环得到目标产物,合成路线如下所示:
[0009][0010]
该路线虽然缩短了反应步骤,但是起始物料5-碘-2,4-二氯嘧啶和丙炔酸甲酯的物料价格较高,再加上丙炔酸甲酯活性较高,在碱作用下也容易发生自身聚合,参与sonogashira偶联反应转化率低,导致路线总收率偏低,成本较高。总之仍需要找到工艺路线简单、收率较高、成本低廉、适宜工业化生产的方法合成曲拉西利关键中间体2'-氯-7',8'-二氢-6'h-螺[环己烷-1,9'-吡嗪并[1',2':1,5]吡咯并[2,3-d]嘧啶]-6'-酮。
技术实现要素:
[0011]
针对现有技术的不足,本发明的目的是提供一种曲拉西利新的中间体及用其制备关键中间体化合物5,具有工艺路线简单、成本低廉、适宜工业化生产等优势。
[0012]
为实现发明目的,本发明采用如下的技术方案:
[0013]
一种曲拉西利中间体化合物3,结构式如下:
[0014][0015]
本发明还涉及所述一种曲拉西利中间体化合物3的合成方法,采用如下的技术方案:
[0016]
一种曲拉西利中间体化合物3的合成方法,包括如下步骤:
[0017]
(1)将起始原料化合物1在钯和铜催化剂和碱作用下与丙炔醇偶联得到中间体化合物2:
[0018][0019]
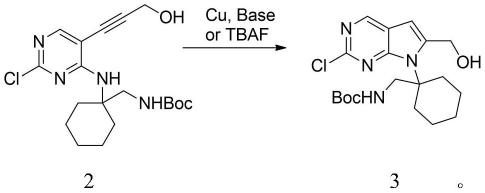
(2)将中间体化合物2在铜盐和碱或tbaf作用下环合得到中间体化合物3;
[0020][0021]
作为优选,所述步骤(1)偶联反应中用到的钯催化剂选自钯碳、醋酸钯、二氯化钯、二氯双(三苯基膦)钯,pd(dppf)cl2或四(三苯基膦)钯;铜催化剂选自碘化亚铜、氯化亚铜或溴化亚铜;不加配体或配体选自三苯基膦、三环己基膦、三叔丁基膦;碱选自三乙胺、二异丙基乙胺、三乙烯二胺、1,8-二氮杂二环[5.4.0]十一碳-7-烯、碳酸钾、碳酸钠或磷酸钾;用到的反应溶剂选自甲醇、乙醇、异丙醇、叔丁醇、叔戊醇、二甲基甲酰胺、二甲基乙酰胺、二甲基亚砜、四氢呋喃、2-甲基四氢呋喃、1,4-二氧六环、乙腈或甲苯;反应温度为0~130℃。
[0022]
作为优选,所述步骤(2)的环合反应选用tbaf作为环合试剂;溶剂选自二甲基甲酰胺、二甲基乙酰胺、二甲基亚砜、二氯甲烷、四氢呋喃、1,4-二氧六环或乙腈;反应温度为25~90℃。
[0023]
作为优选,所述步骤(2)的环合反应选用铜催化剂和碱作用下关环,铜催化剂选自碘化亚铜、氯化亚铜或溴化亚铜;碱选自三乙胺、二异丙基乙胺、三乙烯二胺、1,8-二氮杂二环[5.4.0]十一碳-7-烯、碳酸钾、碳酸钠或磷酸钾;溶剂选自二甲基甲酰胺、二甲基乙酰胺、二甲基亚砜、n-甲基吡咯烷酮或dmpu;反应温度为50~130℃。
[0024]
本发明还涉及用中间体化合物3制备关键中间体化合物5的合成方法,采用如下的技术手段:
[0025]
一种曲拉西利中间体化合物5的合成方法,包括如下步骤
[0026]
(1)将中间体化合物3在氧化剂作用下氧化得到中间体化合物4;
[0027][0028]
(2)将中间体化合物4在酸作用下脱boc并一锅法成环得到中间体化合物5;
[0029][0030]
作为优选,所述步骤(1)的氧化反应选用tempo催化氧化方法进行氧化,其中,氧化剂选自nbs、ncs、二溴海因、醋酸碘苯、次氯酸钠、亚氯酸钠或tcca;不加助剂或选择加入助剂选自碳酸氢钠、氨基磺酸、溴化钠或异丁烯提高转化率;反应溶剂选自四氢呋喃、二氯甲烷、1,2-二氯乙烷、甲苯、乙酸乙酯、乙酸异丙酯或甲基叔丁醚;反应温度为-20~90℃。
[0031]
作为优选,所述步骤(2)的脱boc并一锅法成环反应酸选用盐酸、硫酸、甲基磺酸、对甲苯磺酸或三氟甲磺酸;反应溶剂选自甲醇、乙醇、异丙醇、叔丁醇、叔戊醇、二氯甲烷、四氢呋喃、1,2-二氯乙烷、甲苯、二甲苯、氯苯或三氟甲苯;反应温度为0~150℃。
[0032]
本发明还涉及一种曲拉西利中间体化合物5的合成方法,采用如下的技术方案:
[0033]
一种曲拉西利中间体化合物5的合成方法,其特征在于以n-(1-((叔丁氧羰基)氨基甲基)环己基)-5-溴-2-氯嘧啶-4-胺化合物1为起始原料,与丙炔醇在钯和铜催化下进行sonogashira偶联得到化合物2,然后在铜盐和碱或tbaf作用下完成环合反应得到化合物3,并将伯醇氧化为羧酸得到化合物4,最后一锅法脱去boc并成环得到目标产物2'-氯-7',8'-二氢-6'h-螺[环己烷-1,9'-吡嗪并[1',2':1,5]吡咯并[2,3-d]嘧啶]-6'-酮化合物5。路线如下:
[0034][0035]
本反应关于曲拉西利中间体化合物5的合成方法相对于现有技术来说优化了工
艺,缩短了路线步骤,提高了路线效率,丙炔醇原料廉价易得,并且转化至羧酸步骤简单,显著降低了工艺成本。该路线操作简单,不仅总收率较高,得到的产品纯度也较高,适合放大生产。
具体实施方式
[0036]
下面对本发明的实施例作详细说明,本实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方式和具体的操作过程,但本发明的保护范围不限于下述的实施例。
[0037]
实施例1
[0038][0039]
三口烧瓶中加入n-(1-((叔丁氧羰基)氨基甲基)环己基)-5-溴-2-氯嘧啶-4-胺化合物1(41.97g,100mmol),丙炔醇(11.21g,200mmol),二异丙基乙胺dipea(25.85g,200mmol)和异丙醇(420ml),搅拌均匀后真空切换氮气3次,氮气保护下加入碘化亚铜(190mg,1mmol),二氯双(三苯基膦)钯(702mg,1mmol),加完后升温至55~60℃反应6-8小时,反应结束过滤,旋去大部分异丙醇,加水420ml,打浆,过滤,收集滤饼干燥得化合物2(32.66g,82.7%)。
[0040]1hnmr(500mhz,dmso-d6)δ8.04(s,1h),6.85-7.04(m,1h),5.65-5.77(m,1h),5.32-5.41(m,1h),4.31-4.47(m,2h),3.25-3.44(m,2h),2.04-2.17(m,2h),1.15-1.62(m,17h)。
[0041]
ms(esi)m/z=395.1[m h]
。
[0042]
实施例1中;碱二异丙基乙胺可用三乙胺、三乙烯二胺、1,8-二氮杂二环[5.4.0]十一碳-7-烯、碳酸钾、碳酸钠或磷酸钾代替;溶剂异丙醇可用甲醇、乙醇、叔丁醇、叔戊醇、二甲基甲酰胺、二甲基乙酰胺、二甲基亚砜、四氢呋喃、2-甲基四氢呋喃、1,4-二氧六环、乙腈或甲苯代替;铜催化剂碘化亚铜可用氯化亚铜或溴化亚铜代替;钯催化剂二氯双(三苯基膦)钯可用钯碳、醋酸钯、二氯化钯、pd(dppf)cl2或四(三苯基膦)钯代替;反应过程中可加入三苯基膦、三环己基膦、或三叔丁基膦配体。
[0043]
实施例2
[0044][0045]
三口烧瓶中加入n-(1-((叔丁氧羰基)氨基甲基)环己基)-5-溴-2-氯嘧啶-4-胺化合物1(41.97g,100mmol),丙炔醇(11.21g,200mmol),二异丙基乙胺dipea(25.85g,
200mmol)和乙醇(420ml),搅拌均匀后真空切换氮气3次,氮气保护下加入碘化亚铜(190mg,1mmol),10%钯碳(1064mg,1mmol),加完后升温至55~60℃反应6-8小时,反应结束过滤回收钯碳,旋去大部分乙醇,加水420ml,打浆,过滤,收集滤饼干燥得中间体式2(30.21g,76.5%)。
[0046]
实施例1中;碱二异丙基乙胺可用三乙胺、三乙烯二胺、1,8-二氮杂二环[5.4.0]十一碳-7-烯、碳酸钾、碳酸钠或磷酸钾代替;溶剂乙醇可用异丙醇甲醇、叔丁醇、叔戊醇、二甲基甲酰胺、二甲基乙酰胺、二甲基亚砜、四氢呋喃、2-甲基四氢呋喃、1,4-二氧六环、乙腈或甲苯代替;铜催化剂碘化亚铜可用氯化亚铜或溴化亚铜代替;钯催化剂钯碳可用二氯双(三苯基膦)钯、醋酸钯、二氯化钯、pd(dppf)cl2或四(三苯基膦)钯代替;反应过程中可加入三苯基膦、三环己基膦、或三叔丁基膦配体。
[0047]
实施例3
[0048][0049]
三口烧瓶中加入化合物2(39.49g,100mmol)和n-甲基吡咯烷酮nmp(197ml),搅拌均匀后真空切换氮气3次,氮气保护下加入氯化亚铜(990mg,10mmol)和1,8-二氮杂二环[5.4.0]十一碳-7-烯dbu(3.04g,20mmol),加完后升温至110~120℃反应过夜。反应结束加水395ml淬灭反应,水相用乙酸乙酯(395ml)萃取,合并有机相水洗2次(99ml),硫酸钠干燥,过滤,浓缩至小体积,缓慢加入石油醚,降温至0~5℃打浆,过滤,收集滤饼干燥,得中间体式3(34.67g,87.8%)。
[0050]1hnmr(500mhz,dmso-d6)δ8.83(s,1h),7.42-7.54(m,1h),6.53(s,1h)5.48-5.57(m,1h),4.62-4.72(m,2h),3.75-3.85(m,2h),2.67-2.85(m,2h),1.58-1.80(m,4h),1.37-1.54(m,13h)ppm.
[0051]
ms(esi)m/z=395.1[m h]
。
[0052]
实施例3中,铜催化剂氯化亚铜可用碘化亚铜或溴化亚铜代替;碱1,8-二氮杂二环[5.4.0]十一碳-7-烯可用三乙胺、二异丙基乙胺、三乙烯二胺、碳酸钾、碳酸钠或磷酸钾代替;溶剂n-甲基吡咯烷酮可用二甲基甲酰胺、二甲基乙酰胺、二甲基亚砜或dmpu代替。
[0053]
实施例4
[0054][0055]
三口烧瓶中加入化合物2(39.49g,100mmol)和thf(395ml),加入tbaf三水合物(94.65g,300mmol),加完后升温至55~60℃反应过夜。反应结束加水395ml淬灭反应,水相用乙酸乙酯(395ml)萃取,合并有机相水洗2次(98ml),硫酸钠干燥,过滤,浓缩至小体积,缓
慢加入石油醚(316ml),降温至0~5℃打浆,过滤,收集滤饼干燥,得中间体式3(33.37g,84.5%)。
[0056]
实施例4中,溶剂四氢呋喃可用二甲基甲酰胺、二甲基乙酰胺、二甲基亚砜、二氯甲烷、1,4-二氧六环或乙腈代替。
[0057]
实施例5
[0058][0059]
三口烧瓶中加入3(39.49g,100mmol)和二氯甲烷(395ml),加入tempo(312mg,2mmol),醋酸碘苯(96.63g,300mmol),加完后室温搅拌6~8小时,然后加热升温至38~40℃反应过夜。反应结束浓缩除去部分二氯甲烷,缓慢加入石油醚(316ml),降温至室温打浆,过滤,收集滤饼干燥,得化合物4(36.88g,90.2%)。
[0060]
实施例5中,氧化剂醋酸碘苯可用nbs、ncs、二溴海因、次氯酸钠、亚氯酸钠或tcca代替;反应溶剂二氯甲烷可用四氢呋喃、1,2-二氯乙烷、甲苯、乙酸乙酯、乙酸异丙酯或甲基叔丁醚代替;反应过程中也可加入助剂选自碳酸氢钠、氨基磺酸、溴化钠或异丁烯提高转化率。
[0061]
实施例6
[0062][0063]
三口烧瓶中加入3(39.49g,100mmol),二氯甲烷(197ml),搅拌溶解后置于冰浴冷却,加入碳酸氢钠(16.8g,200mmol),加入tempo(312mg,2mmol),分批次加入二溴海因(60.04g,210mmol),升温至室温反应6-8小时。反应结束加入10%柠檬酸溶液调节ph至3~4,过滤除去部分固体,分液,水相再用二氯甲烷(98ml)萃取1次,合并有机相水(98ml)洗2次,无水硫酸钠干燥,过滤,浓缩,浓缩后加入石油醚打浆,过滤分离出固体,干燥得化合物式4(35.12g,85.9%)。
[0064]
实施例6中,氧化剂二溴海因可用醋酸碘苯、nbs、ncs、次氯酸钠、亚氯酸钠或tcca代替;反应溶剂二氯甲烷可用四氢呋喃、1,2-二氯乙烷、甲苯、乙酸乙酯、乙酸异丙酯或甲基叔丁醚代替;反应过程碳酸氢钠可不加,或者也可用氨基磺酸、溴化钠或异丁烯代替来提高转化率。
[0065]
实施例7
[0066]
[0067]
三口烧瓶中加入2(40.89g,100mmol)和甲苯(409ml),加入对甲苯磺酸一水合物(41.85g,220mmol),加热至回流带水反应8~10小时,反应结束冷至室温,加入5%碳酸氢钠水溶液(205ml)搅拌分液,有机相水洗1次,收集有机相蒸出大部分甲苯,缓慢加入石油醚(245ml),缓慢降温至0~5℃打浆,过滤,收集滤饼干燥,得目标产物式5(33.37g,88.1%)。
[0068]
实施例7中,甲基磺酸可用盐酸、硫酸、对甲苯磺酸或三氟甲磺酸代替;反应溶剂甲苯可用甲醇、乙醇、异丙醇、叔丁醇、叔戊醇、二氯甲烷、四氢呋喃、1,2-二氯乙烷、二甲苯、氯苯或三氟甲苯代替。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。