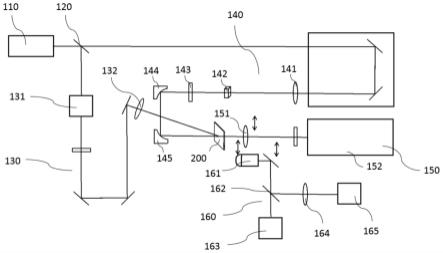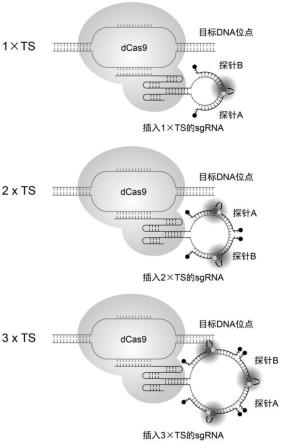
一种基于crispr/dcas9系统与寡核苷酸探针的活细胞dna标记信号放大方法
技术领域
1.本发明涉及在活细胞中荧光标记特定dna位点的成像技术,具体涉及一种基于crispr/dcas9系统与寡核苷酸探针的活细胞dna标记信号放大方法。
背景技术:
2.研究特定dna的时空动态行为,并完全掌握其与细胞生理和疾病发生发展的关系,对于人类健康的发展具有十分重要的意义。荧光原位杂交技术(fluorescence in situ hybridization,fish)是当前被用于dna成像的主要手段,该技术利用多个带有荧光标记的寡核苷酸探针靶向同一个dna位点,实现对特定dna位点的荧光标记与显微成像。尽管fish已帮助研究者在dna研究领域取得了前所未有的进展,但它也存在无法回避的局限性,例如,它需要对细胞进行固定处理,因此无法在活细胞生理条件下对细胞内的dna进行直接观察和研究,也不适用于精细的时程监测。基于成簇规律间隔短回文序列(clustered regularly interspaced short palindromic repeats,crispr)/crispr相关(crispr-associated,cas)系统的活细胞dna标记方法是解决这一问题的有效手段。crispr/cas系统可通过cas蛋白酶与单链向导rna(single-guide rna,sgrna)所形成的cas9-sgrna复合体对具有特定序列的双链dna进行切割。进一步研究表明,失去切割活性的cas9蛋白突变体(即nuclease-deactivated cas9,dcas9)仍然具备与sgrna形成复合体靶向特定dna序列的能力,推动了基于crispr/dcas9系统标记特定dna位点的研究领域(以下简称“crispr成像系统”)的开创与发展。现阶段大多数crispr成像系统借助被荧光蛋白标记的dcas9-sgrna复合体实现对特定dna位点的成像,然而,荧光蛋白具有分子量大、亮度较低、易被光漂白等缺点,在高灵敏度、长时程追踪dna位点中的应用能力有限。化学小分子荧光团(有机染料)具有比荧光蛋白更低的分子量、更高的亮度以及更强的光稳定性,具备高灵敏度、长时程追踪dna位点的潜力。然而利用有机染料对dcas9或sgrna进行特异性标记具有一定的难度。xiaotian wu等(nucleic acids res.2018;46:e80)于2018年报道了一种将携带有机染料的寡核苷酸探针(分子信标,molecular beacon,mb)与crispr/dcas9系统相结合的新型dna标记方法,名为crispr/mb,该方法通过给sgrna插入一段能够被mb特异性识别的序列(target sequence,ts)使得结合于目标dna位点的dcas9-sgrna复合体能够被mb特异性标记,从而“点亮”目标dna位点。实验证实,在对人类基因组端粒(telomere)高重复序列的成像上,crispr/mb具有比基于荧光蛋白的标记方法更高的标记能力与更强的光稳定性。之后,在crispr/mb成像平台的基础上,shiqi mao等(nucleic acids res.2019;47:e131)对sgrna进行了进一步改造,使得被改造后的sgrna的ts序列能够与一对可发生荧光共振能量转移(fluorescence resonance energy transfer,fret)的供体/受体mb互补,发展出一种名为crispr/dual-fret mb的dna标记方法。由于只有当两个mb同时互补于同一个sgrna时,供体mb的荧光团才能与受体mb的荧光团相互靠近并发生fret,该方法能够更好地区分来自基因组位点的信号与游离mb造成的背景荧光。实验证实,crispr/dual-fret mb具有比
crispr/mb(基于单个mb)更高的灵敏度。
3.为了进一步提高标记效率,一个有效的解决方案是增加标记单个dna位点的有机染料数目来实现标记信号强度的放大。尽管可以通过增加sgrna的数目来实现这一目标,单个dna位点上结合过多的dcas9-sgrna复合体可能会对该位点的运动造成影响,从而无法反映其真实的动态行为。此外,由于dna位点上并非每个区域都能够充分被sgrna识别,增加标记所需的sgrna数目也可能增大标记难度。
技术实现要素:
4.针对上述问题,本发明通过增加单个sgrna所携带的有机染料数目对现有的crispr/mb平台进行改造,旨在不增加用于标记单个dna位点的dcas9-sgrna复合体数目的同时实现有效的信号强度放大,并为基于crispr/dcas9系统与寡核苷酸探针的活细胞dna标记方法提供一种通用的信号放大方法。
5.本发明提供的技术方案的策略如下:
6.本发明方案使用crispr/dual-fret mb作为基于crispr/dcas9系统与寡核苷酸探针的活细胞dna标记方法的代表。基于单个mb的crispr/mb方法以及基于crispr与其他寡核苷酸探针的dna标记方法理论上均可以使用本发明的方法进行改造。本方案中,实现增加单个sgrna上所携带的有机染料数目需要运用分子合成技术增加sgrna上所插入的ts数目。前人的研究结果表明,sgrna的tetraloop、stem-loop 2、3
’‑
tail region等区域均具有高度可改造性,本方案选择在stem-loop 2区域进行改造,其他区域理论上也可以使用本发明的方法进行改造。为评估增加ts数目的效果,本方案给sgrna分别插入串联重复2次和3次的ts序列(2
×
ts和3
×
ts),以在sgrna的stem-loop 2区域插入1次ts序列(1
×
ts)作为对照,并对这些sgrna标记同一dna位点的信号强度进行比较。携带串联重复更高次数ts序列的sgrna理论上也可使用本发明提供的方案进行活细胞dna标记。具体方案如下:
7.一种基于crispr/dcas9系统与寡核苷酸探针的活细胞dna标记信号放大方法,包括:
8.(1)构建靶向目标dna位点的sgrna表达片段,所述sgrna表达片段在sgrna的stem-loop 2区域插入串联重复多次的ts序列,所述ts序列由能够被探针a识别的3’端序列tsa和能够被探针b识别的5’端序列tsb组成;
9.(2)构建能够发生fret的供体mb和受体mb:合成序列与tsa特异性互补的寡核苷酸探针a和序列与tsb特异性互补的寡核苷酸探针b,在探针a的3’端连接供体荧光团或受体荧光团,对应地在探针b的5’端连接受体荧光团或供体荧光团;带有供体荧光团的探针为供体mb,带有受体荧光团的探针为受体mb,即探针a作为供体mb时,探针b作为受体mb,而探针a作为受体mb时,探针b作为供体mb;
10.(3)将步骤(1)构建的sgrna表达片段与dcas9蛋白表达片段克隆到质粒表达载体中,并将该质粒表达载体转染到活细胞中;
11.(4)将步骤(2)构建的供体mb和受体mb通过电转的方式共同导入步骤(3)转染后的活细胞中;
12.(5)对电转后的活细胞进行荧光显微镜成像,得到放大的目标dna位点的荧光信号强度。
13.上述步骤(1)中,ts序列的串联重复次数优选为2-3次。所述ts序列是所述细胞基因组中不存在的无义序列,以避免内源核酸分子对成像造成干扰。ts序列的长度优选为40~70核苷酸(nucleotide,nt),其中tsa和tsb的长度优选均为19~34nt。在串联的两个ts序列之间插入一段间隔序列,其目的为减少结合于相邻ts序列上的探针a与探针b之间的空间位阻,无特定的碱基序列要求,此外为了尽量降低dcas9-sgrna复合体对于被标记的dna位点运动的影响,间隔序列不宜过长,优选为2~4nt。
14.在本发明的一个实施例中,所述ts序列的编码序列为:5
’‑
caggagttgtgtttgtggacgaagcaagctcagtcacgacatcacttacgct-3’(seq id no:1)。相应的,所述探针a的序列为:5
’‑
cucagcguaagugaugucgugacugag-3’(seq id no:2);所述探针b的序列为:5
’‑
cuucguccacaaacacaacuccugaag-3’(seq id no:3)。其他已验证的ts序列的编码序列例如:1)5
’‑
caggagttgtgtttgtggacgaagcaagctagcgcgaggatagtgatttagagc-3’(seq id no:4);2)5
’‑
caggagttgtgtttgtggacgaagcaagggtcgaaccagtggaacctacaacg-3’(seq id no:5)。此外,其他在目标细胞基因组中无同源序列且可被寡核苷酸探针特异性识别的ts序列同样可应用于本发明。
15.上述步骤(2)中,所述探针a和探针b为rna探针,为了使探针在活细胞环境中能够有效抵抗核酸酶的降解,可以对探针序列进行化学修饰。能够抵抗核酸酶降解的化学修饰包括但不限于2
’‑
o-甲基化(2
’‑
o-methyl)修饰、硫代磷酸(phosphorothioate,ps)修饰、锁核酸(locked nucleic acid,lna)修饰、吗啉基(morpholino)修饰等。可以对组成探针的所有或部分核糖核苷酸进行一种或多种化学修饰。
16.连接在探针a和探针b上的供体荧光团和受体荧光团组成fret荧光对,可以使用现有技术中已证实fret效率较高的fret荧光对,例如atto550/atto647n、atto488/atto550、alexa546/alexa647、cy3/cy5等荧光对。
17.进一步地,步骤(2)可以在所述探针a的5’端连接淬灭基团,在所述探针b的3’端连接淬灭基团。
18.上述步骤(3)中,将质粒表达载体通过转染试剂如6等转染到活细胞中。优选的,转染24小时后再进行步骤(4)的电转。
19.步骤(5)在电转后24小时活细胞进行荧光显微镜成像,通过计算并比较被携带不同串联重复次数ts序列的sgrna标记的目标dna位点的荧光信号强度,可以发现相对于插入1个ts序列,串联重复2次和3次ts序列信号强度显著增大。
20.本发明的有益效果主要体现在:在不增加用于标记单个dna位点的dcas9-sgrna复合体数目的同时实现了有效的信号强度放大,为基于crispr/dcas9系统与寡核苷酸探针的活细胞dna标记方法提供一种通用的信号放大方法。
附图说明
21.图1.使用携带不同串联重复次数ts序列的sgrna标记目标dna位点示意图。
22.图2.使用携带不同串联重复次数ts序列的sgrna标记人类基因组端粒高重复序列的荧光成像与信号分析结果,其中:a为宽场显微镜荧光成像效果图,比例尺=10μm;b为被标记的端粒的荧光信号强度,图中数值代表平均值
±
标准误,*代表显著性差异(单因素anova分析,*代表p《0.01,**代表p《0.001)。
具体实施方式
23.下面的实施例以人类基因组端粒作为模式dna位点对本发明所提供的信号放大方法进行说明。
24.本实施例通过分子合成技术得到了3种靶向人类基因组端粒高重复序列的sgrna(sgtelo),这3种sgrna的stem-loop 2区域分别携带串联重复1次、2次或3次的ts序列(sgtelo_1
×
ts、sgtelo_2
×
ts或sgtelo_3
×
ts),该ts序列由tsa(可被连接着atto550荧光团的供体mb特异性识别)和tsb(可被连接着atto647n荧光团的受体mb特异性识别)组成,两对mb/ts之间无交叉干扰。当供体mb与受体mb同时结合于同一个ts序列上,供体mb与受体mb的荧光团会相互靠近发生fret,本实施例即通过比较被携带不同串联重复次数ts序列的sgrna标记的端粒的fret信号强度评估本方案的信号放大效果。具体实施方式如下:
25.1试剂与仪器
26.1.1主要试剂与材料
27.1)靶向人类基因组端粒高重复序列且在stem-loop 2区域分别携带串联重复1次、2次或3次的ts序列的sgrna表达载体(可交由苏州金唯智生物科技有限公司合成,sgrna具体序列见表1);
28.2)质粒表达载体sgrna/egfp/pdcas9-c1(原始文献为nucleic acids res.2018;46:e80;该载体在本实施例中被用于构建可同时表达sgrna、dcas9蛋白以及转染指示蛋白egfp的质粒表达载体,构建方法见实验方法2.1)。
29.3)mb(可交由integrated dna technologies公司合成):anti-mtsa mb(供体mb,3’端连接着atto550荧光团);anti-mtsb mb(受体mb,5’端连接着atto647n荧光团)。
30.4)相关限制性内切酶、t4连接酶(可购于new england biolabs公司)。
31.5)感受态大肠杆菌细胞。
32.6)质粒提取试剂盒(可购于omega bio-tek公司)。
33.7)人胚胎肾细胞293(hek293)。
34.8)含10%(vol/vol)胎牛血清(可购于pan
tm biotech公司)和1
×
glutamax
tm
(可购于thermo fisher公司)的dmem培养基(可购于corning公司);10
×
pbs(可购于corning公司);胰蛋白酶(可购于thermo fisher公司)。
35.9)转染试剂6(可购于promega公司)。
36.1.2主要仪器
37.1)pcr仪、凝胶电泳仪、生化培养箱、摇床、恒温孵化器。
38.2)细胞培养箱、生物安全柜。
39.3)转染系统。
40.4)荧光显微镜。
41.2实验方法
42.2.1质粒构建
43.分别以交由公司合成得到的3种sgrna表达载体为模板,利用引物对(正向引物:5
’‑
tccaaactcatcaatgtatcttattagagggcctatttcccatgattcc-3’,seq id no:6;反向引物:5
’‑
atgaccccgtaattgattactattatgagcggataacaatttcacac-3’,seq id no:7)pcr扩增出u6启动子-sgrna表达片段,然后将扩增产物利用gibson assembly克隆方法插入被asei酶切
后的质粒表达载体sgrna/egfp/pdcas9-c1中(asei酶切用于去除原载体上的u6启动子-sgrna表达片段),最终得到本实施例需要用到的3种质粒表达载体:sgtelo_1
×
ts/egfp/pdcas9-c1;sgtelo_2
×
ts/egfp/pdcas9-c1;sgtelo_3
×
ts/egfp/pdcas9-c1。
44.2.2细胞转染与电转
45.当六孔板中的细胞生长至50%-70%覆盖率时,使用转染试剂6分别将实验方法2.1中所述的3种质粒表达载体转染进入hek293细胞,质粒转染量均为2μg。
46.转染后24小时,使用转染系统将供体mb与受体mb(各1μm)共电转进细胞中。
47.2.3荧光成像与结果分析
48.1)电转24小时后,使用奥林巴斯倒置荧光显微镜(olympus ix83)进行荧光成像,并使用成像软件cellsens dimension获取细胞图像(参数设置:曝光时间设为500ms,电子倍增增益(em gain)设为30)。
49.2)荧光成像结果如图2中a所示。在转染3种质粒表达载体的hek293细胞中,细胞核中的fret荧光信号形态相似,均呈现为较小的荧光亮点。不同的是,随着所使用的sgrna上ts的串联重复次数的增加,荧光亮点的亮度也有所增加。荧光成像结果提示,ts重复次数的增加对信号起到了一定的放大效果。
50.3)对细胞核内荧光亮点的信号强度进行定量分析。使用图像处理软件imagej打开细胞图像,首先用subtract background工具减去背景荧光,再根据荧光亮点的情况用threshold工具设置恰当的阈值将亮点与背景区分开,通过analyze particles工具测量得到亮点的荧光信号强度。分析结果如表2及图2中b所示。在本实施例中,随着所使用的sgrna上ts的串联重复次数的增加,荧光亮点的信号强度显著增加,证实增加ts重复次数可以实现对信号的有效放大。
51.表1.本实施例所使用的sgrna序列
[0052][0053]
表中加着重号部分为sgrna的spacer区域(用于识别目标dna位点),下划线部分为ts序列(用于结合供体/受体mb)。
[0054]
表2.端粒荧光信号强度分析结果
[0055]
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。