1.本发明涉及抗原预测技术领域,尤其涉及一种预测免疫原性的模型和应用。
背景技术:
2.肿瘤特异性的人类白细胞抗原(hla)肽只在癌细胞表面表达,是免疫系统区分癌细胞和正常细胞的理想目标。然而由于t细胞的高特异性,这些多肽只有一小部分能够被t细胞识别,从而触发免疫反应,这类具有抗原性的hla肽被称为肿瘤新抗原。尽管新抗原在癌症免疫治疗中有很好的作用,但新抗原的发现几率非常低,通常每个患者在数千个体细胞突变中发现不到6个。常用的新抗原的发现方法(如cn111415707a)是对每个患者个体肿瘤基因组进行高通量的dna序列检测,以寻找所有的肿瘤特异性突变;然后对这些突变表达出来的蛋白和多肽进行抗原性预测,将潜在的新抗原肽段进行体外合成,然后再进行体外验证,这些技术不仅费时而且非常昂贵。
3.免疫原性预测是使用生物信息工具预测hla-i和hla-ii(phla)复合物的结合亲和力,或者整个phla呈递途径。由于每个病人的癌症和免疫治疗都与大量的个体基因突变相关,hla复合体、t细胞群以及肿瘤突变本身在不同患者之间存在显著差异,因此,癌症免疫治疗应该充分考量个性化方法。一种潜在的方法(如cn113160887a)是对患者的tcr进行测序,并根据其序列或结构预测tcr-phla识别。然而,尽管对tcr进行了持续而密集的测序,但很难对每个患者完整的tcr序列进行测序。因此,在不对tcr进行测序的情况下,建立一个预测tcr-phla识别的个性化模型,是急需解决的难题。
技术实现要素:
4.有鉴于此,本发明提出了一种预测免疫原性的模型和应用,通过模拟个体cd8 t细胞中枢耐受性预测该患者候选新抗原的免疫原性。
5.本发明的技术方案是这样实现的:一方面,本发明提供了一种预测免疫原性的模型,该模型的构建包括如下步骤:
6.s1:提取患者的hla-i多肽,进行ms数据采集,然后将采集到的ms数据在swiss-prot人类蛋白数据库进行搜索,对鉴定出的hla-i多肽进行特征长度分布检查;将长度小于8或者大于14的肽段剔除掉,得到的肽段为患者的阴性选择数据模型;
7.s2:下载iedb t细胞表位数据库,选择与患者的hla-i等位基因相匹配的阳性表位,剔除长度小于8或者大于14的肽段,得到患者的阳性选择数据模型;
8.s3:采用tensorflow将氨基酸字母转换为整数索引,使用步骤s1的阴性选择数据模型和s2的阳性数据模型训练keras序列模型,并进行100轮训练,最后仅保留了验证损失最好的模型;
9.s4:将每个患者的训练数据分成训练集、验证集和测试集进行测试,并为每个患者训练100个集成模型,根据集成模型在验证集上的表现对其进行排序,并选择前10个模型的平均值作为该患者的最终预测模型;
10.s5:根据最终预测模型输出的肽段进行打分,得分越高表示输入肽更有可能被cd8 t细胞识别。
11.在以上技术方案的基础上,优选的,步骤s1中,使用peaks xpro将ms数据在swiss-prot人类蛋白数据库中进行搜索。
12.在以上技术方案的基础上,优选的,步骤s2中,下载iedb t细胞表位数据库时,设置如下过滤条件:线性表位,hla-i类,宿主是人类或老鼠。
13.在以上技术方案的基础上,优选的,步骤s2中所述阳性表位包括阳性、阳性-高、阳性-中间值和阳性-低表位。
14.在以上技术方案的基础上,优选的,步骤s3,tensorflow中keras序列模型的训练包括:一个包含8个神经单元的嵌入层,一个包含8个单元的双向lstm层,一个带有l2正则化的全连接层和一个sigmoid激活层。
15.在以上技术方案的基础上,优选的,步骤s3,使用adam优化器和二元交叉熵函数进行100轮训练。
16.在以上技术方案的基础上,优选的,步骤s4中,训练集、验证集和测试集的比例为8:1:1。
17.在以上技术方案的基础上,优选的,步骤s4中,由于阴性多肽的数量通常比阳性多肽的数量高,因此对阴性肽进行降采样,保证数据集中阳性肽与阴性肽的比例相同。
18.另一方面,本发明提供了一种预测免疫原性的模型在预测癌症患者候选新抗原免疫性中的应用。
19.本发明的一种通过模拟个体cd8 t细胞中枢耐受性预测免疫原性的模型和应用相对于现有技术具有以下有益效果:本发明使用来自患者hla-i自身肽的ms免疫肽组数据的作为阴性训练数据,使用来自iedb的等位基因匹配的阳性t细胞表位作为阳性训练数据。多次实验表明,本发明的预测模型在个体患者预测新抗原的准确率达到了79%,优于现有的免疫原性预测工具。更重要的是,能够按照免疫原性强弱对候选肽进行排名,这些具有免疫原性的候选肽排列在前2%,从而大大节省了进一步体外验证的试验成本和时间成本。
附图说明
20.为了更清楚地说明本发明实施例或现有技术中的技术方案,下面将对实施例或现有技术描述中所需要使用的附图作简单地介绍,显而易见地,下面描述中的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下,还可以根据这些附图获得其他的附图。
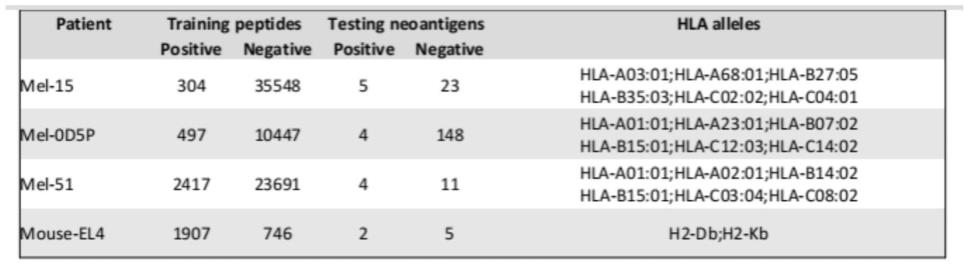
21.图1为具体实施中3例癌症患者和1个小鼠癌细胞系hla等位基因、训练和评估数据汇总;
22.图2为具体实施中预测工具对实验验证的新抗原的roc曲线下面积;
23.图3为具体实施中通过对患者mel-15的ms数据从头测序确定的候选多肽中免疫原性新抗原的预测排名。
具体实施方式
24.下面将结合本发明实施方式,对本发明实施方式中的技术方案进行清楚、完整地
描述,显然,所描述的实施方式仅仅是本发明一部分实施方式,而不是全部的实施方式。基于本发明中的实施方式,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施方式,都属于本发明保护的范围。
25.本发明采用的技术方案是,通过建立cd8阳性选择和阴性选择模型,即t细胞的中枢耐受的模型,来预测癌症患者cd8 t细胞的候选新抗原的免疫原性,该模型的构建包括如下步骤:
26.s1,提取患者的hla-i多肽,然后进行ms数据采集。
27.s2,使用peaks xpro将ms数据与标准的swiss-prot人类蛋白数据库进行检索无酶特异性切割,假阳性率(fdr)为1%。
28.s3,对鉴定出的多肽进行hla-i特征长度分布检查;将长度《8或》14的氨基酸多肽剔除掉,得到的多肽被认为是患者的hla-i自身多肽,这些多肽是不被患者的t细胞识别(即非免疫原性),通过此方法筛选的肽段为患者的阴性选择数据模型。
29.s4,下载iedb t细胞表位数据库,设置如下过滤条件:线性表位,hla-i类,宿主为人类或老鼠。
30.s5,选择与患者的hla-i等位基因相匹配的阳性表位,阳性表位包括:阳性、阳性-高、阳性-中间值和阳性-低;剔除长度《8或》14的氨基酸表位的多肽,由此产生的多肽被假定为被患者的t细胞识别(即免疫原性),通过此方法筛选的肽段为患者的阳性选择数据模型。
31.s6,在tensorflow中将氨基酸字母转换为整数索引,肽序列用0-15表示,使用步骤s3的阴性数据模型和步骤s5阳性数据模型来训练一个二元分类模型。tensorflow中keras序列模型的训练包括:一个包含8个神经单元的嵌入层,一个包含8个单元的双向lstm层,一个带有l2正则化的全连接层和一个sigmoid激活层。并使用adam优化器和二元交叉熵损失进行100轮训练,最后仅保留了验证损失最好的模型。
32.s7,将每个患者的训练数据分成三组进行测试:训练集、验证集和测试集,训练集:验证集:测试集的比例为8:1:1,三组中没有重叠的多肽。由于阴性多肽的数量通常比阳性多肽的数量高出几倍,因此对阴性肽进行了降采样,保证数据集中阳性肽与阴性肽的比例相同,并为每个患者训练了100个集成模型。根据集成模型在验证集上的表现对其进行排序,并选择前10个模型的平均值作为该患者的最终预测模型。
33.s8,该免疫原性预测模型被命名为deepimmun。根据最终预测模型输出的肽段进行打分,分数从0到1,得分越高表示输入肽更有可能被特定癌症患者的cd8 t细胞识别。
34.具体实施方式中,分别对3例癌症患者和1个小鼠癌细胞系(mel-15,mel-0d5p,mel-51,and mouse-el4)的hla ms数据进行建模,每个患者的hla-i自身多肽数量为746-35548个,而免疫原性表位数量为304-2417个,见图1。
35.为了进行性能评估,我们将deepimmun与三种领先的工具进行了比较,包括prime、netmhcpan和iedb免疫原性预测器。netmhcpan是最早和最常见的进行hla-i预测的工具之一。iedb免疫原性预测因子是最早的hla-i免疫原性预测工具,它显示了免疫原性与氨基酸位置4-6之间的关联,以及氨基酸残基的其他特性,如疏水性、极性或大的芳香侧链。prime是一种最新的免疫原性预测工具,可以同时模拟hla-i结合和tcr识别。值得注意的是,netmhcpan是被设计用于预测hla-i结合,而不是免疫原性,但它被广泛应用于许多新抗原
预测工作流程。本发明基于两个标准评估了这四种预测工具。
36.第一个标准是他们对候选新抗原的预测的受试者工作特征曲线(roc-auc)下的面积。deepimmun在meml-15和mel-0d5p患者上的表现优于其他工具。在患者mel-51上,deepimmun、prime和netmhcpan表现相当。对于mouse-el4,iedb预测器达到了最高的auc为90%,其次是deepimmun和netmhcpan(prime不支持小鼠数据),见图2。总的来说,deepimmun在每个数据集上的平均auc为79%。四种预测工具之间的相对性能也反映了其基础模型的特征:deepimmun是一个个性化模型;prime是等位基因特定模型;iedb预测器是在有限数据集上训练的一般模型;netmhcpan是hla结合的预测模型。
37.第二个评估标准是对从患者中识别出的突变多肽中的潜在新抗原进行排序的能力。这些突变的多肽,包括新抗原,不能在标准的swiss-prot人类蛋白数据库中找到。因此,首先将测序工作流程应用于患者的ms数据,以识别数据库中不存在的新肽段。然后应用这四种预测工具(deepimmun、prime、netmhcpan和iedb)对这些候选肽进行排序。新抗原的顺序在候选肽列表越靠前说明模型的准确性越高。我们基于ms的方法只考虑了从ms数据中识别出的候选肽,而其他基因组方法通常考虑了从基因组序列中翻译出来的候选肽。
38.对患者mel-15肿瘤组织的ms数据进行从头测序确定的3638个候选肽中的5个新抗原进行排序,结果见图3。5个新抗原中的前两种新抗原klilwrglk和rlflglaik被deepimmun排在前1%以内,而接下来的两种新抗原griafflky和rtyslssalr分别排在前1.5%和15%以内。最后一种新抗原sqiilrqh在4中四种预测工具的排名都很低,但是它已由willelm等人鉴定和检测。总的来说,deepimmun在这个排名评估中优于其他工具。
39.以上所述仅为本发明的较佳实施方式而已,并不用以限制本发明,凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。