1.本发明属于分析化学领域,具体涉及一种检测草甘膦的荧光试纸及其制备方法与应用。
2.
背景技术:
3.草甘膦(n-(磷酰甲基)甘氨酸,glyphosate)是一种非选择性广谱有机磷农药,具有高极性、低挥发性等特点,其作用机制就是借助其在植物体内优秀的内吸传导性来抑制植物体正常的生长过程,即通过抑制芳香族氨基酸和其他化学物质的合成,进而影响植物的光合作用和其他相关生化途径的作用,被广泛应用于农业、林业、城市规划和家用的除草和植被控制。草甘膦主要应用于农业、畜牧业和种植业中。相较于其他除草剂而言,草甘膦的价格低廉,深受喜爱。不同作物中的草甘膦用量存在差异,如大豆可达4.6
×
10
7 kg/年,玉米可达2.6
×
10
6 kg/年,棉花达4.8
×
10
6 kg/年。目前全球草甘膦的年产量约为8.25
×
10
8 kg(do et al., 2015; santos, pontes, santiago, fiorucci, & arruda, 2020; saunders & pezeshki, 2015)。
4.过去研究认为,虽然草甘膦对鱼类、蚤类、藻类的急性毒性均属于低毒,对哺乳动物急性毒性更低,但是由于草甘膦的使用量迅速攀升,在土壤、地表水和地下水中残量激增,严重超出了土壤吸附降解能力,人们通常可以在陆地水和海洋水等环境中检测到草甘膦,进而会污染食品原材料以及食品,对人类健康造成一定程度的威胁。新的研究表明,草甘膦具有内分泌毒性、神经毒性和细胞毒性,严重影响人类的心血管系统,在2015年,世界卫生组织国际癌症研究机构将其列为“可能对人类致癌”,在欧洲水框架指令的2008/105/ec指令附件iii中,草甘膦被视为“优先危险物质”,因此,开发新技术以检测草甘膦的残留迫在眉睫(gotti, fiori, bosi, & dinelli, 2019; rigobello-masini, pereira, abate, & masini, 2019; fares zouaoui et al., 2020)。
5.目前,草甘膦的检测主要依赖于仪器分析方法,比如毛细电泳检测法、高效液相色谱法、电化学检测法、红外光谱法、化学分析法、免疫分析法等。这些方法的样品前处理复杂,操作步骤繁琐,时间较长,而且需要专业人员进行仪器的维护和操作,导致检测成本高,难以实现现场快速检测。通过调查发现,市面上不依赖仪器的草甘膦检测产品很少,其中较为常见的是美国abraxis草甘膦检测试剂盒(现已停产),其原理是酶联免疫法,产品需冷藏保存,冷链运输,检测成本高,易发生交叉反应影响测量的灵敏度且检测条件较为苛刻;还有上海飞测公司的草甘膦荧光定量快速检测卡,采用荧光侧向免疫层析(fluorescent lateral flow,flf)方法学原理,但试纸价格较高。因此,发展简单快速、低成本、运输方便的草甘膦分析检测技术能够更好地监测和分析其在植物源性食品中的污染现状,以保障我国食品安全和人民健康生活((ding & yang, 2013; sanch
í
s et al., 2012; viirlaid et al., 2019; f. zouaoui et al., 2022)。
6.
技术实现要素:
7.发明目的:针对现有酶联免疫法快速测定草甘膦存在的问题,本发明提供了一种检测草甘膦的荧光试纸及其制备方法与应用,该荧光试纸为一种用于快速检测有机磷农药残留的试纸光学传感器,该光学传感器利用分子印迹技术制备而成,能够特异性识别食品中的有机磷,具有良好的重现性以及稳定性,有效解决了样品预处理复杂,以及现有技术产品成本高、检测环境受到限制的问题。
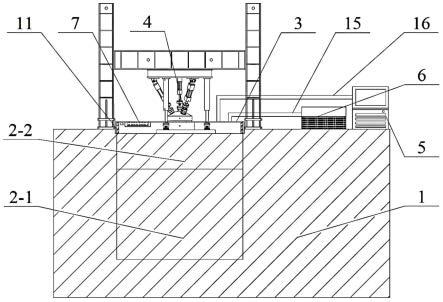
8.技术方案:一种检测草甘膦的荧光试纸的制备方法,步骤如下:步骤一.分子印迹聚合物的制备:将草甘膦模板和功能单体加入溶剂中搅拌,然后加入荧光单体进行预组装,再加入四乙氧基硅酸酯搅拌,离心后经水、乙醇、磷酸盐洗涤后去除草甘膦模板,获得对草甘膦具有特殊结合位点的分子印迹聚合物(mip),其中所述功能单体为苯硼酸硅烷化试剂,所述荧光单体为荧光硅烷试剂,所述荧光硅烷试剂为功能单体和异硫氰酸酯荧光素的结合物,所述溶剂为纯水/无水乙醇的共溶剂;步骤二.共价修饰策略构筑分子印迹试纸:利用硅胶离子中残留的活性基团,通过共价键反应将步骤一制备的分子印迹聚合物固载至试纸上,获得检测草甘膦的荧光试纸。
9.作为优选,所述步骤一中苯硼酸硅烷化试剂为3-氨丙基三乙氧基硅烷(aptes)、n-[3-(三甲氧基硅基)丙基]乙二胺(aaptms)或3-[2-(2-氨基乙基氨基)乙基氨基]丙基-三甲氧基硅烷(aaaptmes),所述荧光硅烷试剂为对应苯硼酸硅烷化试剂与异硫氰酸酯荧光素(fitc)反应获得。
[0010]
作为优选,所述步骤一中草甘膦模板的量与溶剂的比值为0.1 mmol:32 ml,功能单体与草甘膦模板的比值为0.6 mmol:0.1 mmol,荧光单体与草甘膦模板的比值为2 μmol:0.1 mmol;四乙氧基硅酸酯与草甘膦模板的比值为3 mmol:0.1 mmol;所述溶剂中纯水和无水乙醇的比值为20 ml:12 ml。
[0011]
作为优选,所述步骤二中共价修饰策略构筑分子印迹试纸的制备方法具体步骤如下:首先将滤纸裁成直径为2.5 cm的圆形得到试纸,使用3-氨丙基三乙氧基硅烷(aptes)在试纸上修饰氨基,再使用戊二醛溶液在试纸上修饰醛基,在步骤一制备聚合物的基础上加入制备好的试纸,搅拌反应4 h后,获得检测草甘膦的荧光试纸。
[0012]
基于上述一种检测草甘膦的荧光试纸的制备方法制备的检测草甘膦的荧光试纸。
[0013]
基于上述检测草甘膦的荧光试纸在测定草甘膦选择性吸附中的应用。
[0014]
基于上述检测草甘膦的荧光试纸在定量检测分析草甘膦中的应用。
[0015]
作为优选,所述定量检测分析步骤如下:利用检测草甘膦的荧光试纸选择性吸附样品中的草甘膦,经过清洗后,测定其荧光信号值,同时测定空白荧光信号值,根据荧光信号差值采用标准曲线法进行草甘膦的定量分析。
[0016]
作为优选,利用检测草甘膦的荧光试纸选择性吸附样品中的草甘膦时将样品滴加到试纸中心位置,产生咖啡圈效应,将样品滴加到试纸中心位置,液体会扩散,其中的草甘膦会被吸附在中心点,其他干扰物会随着咖啡圈效应扩散到试纸边缘。
[0017]
本发明所述检测草甘膦的荧光试纸是将能够特异性识别草甘膦的分子印迹聚合物结合在试纸上实现快速检测。利用分子印迹技合成产生特定的分子空穴,模拟抗原-抗体
的相互作用以达到特异性识别的目的。
[0018]
有益效果:与现有技术相比,本发明具有如下优点:1.本发明使用预先制备好的分子印迹聚合物,对草甘膦具有特异性;2.原位聚合法能够使聚合物结合更牢固,避免分子识别材料的泄露,该方法能显著提高传感分析的准确度和重现性;3.试纸光学传感器操作简单,试剂用量少,分析时间较短,大大的提高了检测的效率以及检测的灵敏性,易于实现现场取样分析。
[0019]
4.相较于酶联免疫法,分子印迹技术具有抗干扰性能强、成本低、制备简单等优点。
[0020]
5. 本发明采用咖啡圈试纸设计,利用咖啡圈试纸的扩散效应、富集效应缓解试纸输送量低的问题,解决样品基质复杂和检测灵敏度低的问题,增加检测的准确性和材料的利用率。
[0021]
综上,本发明所述检测草甘膦的荧光试纸的特点在于将能够特异性识别草甘膦的荧光分子印迹聚合物固载在试纸上,用于特异性识别草甘膦。该特异性识别能力依赖于利用分子印迹技术合成产生的分子印迹空腔,其能够模拟抗原-抗体的相互作用以达到特异性识别的目的,在试纸上修饰氨基和醛基以实现荧光分子印迹聚合物的固载,以光学仪器作为数据分析工具实现了对草甘膦的定量检测。
[0022]
附图说明
[0023]
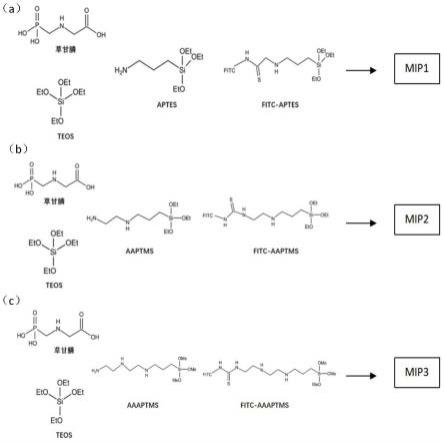
图1. 咖啡圈试纸的制备和使用示意图;图2. 三种分子印迹聚合物的合成路线图,图中(a)为mip1的合成路线图,(b)为mip2的合成路线图,(c)为mip3的合成路线图;图3. mip1(a)、nip1(b)、mip2(c)、nip2(d)、mip3(e)和nip3(f)的扫描电镜图像;图4. 不同浓度mip1及nip 1荧光响应变化率曲线(激发波长471 nm,发射波长517 nm);图5. 不同浓度mip2及nip 2荧光响应变化率曲线图(激发波长471 nm,发射波长517 nm);图6. 不同浓度mip3及nip 3荧光响应变化率曲线图(激发波长471 nm,发射波长517 nm);图7. 草甘膦荧光分子印迹纳米粒子对不同农药的响应分析(mip1、nip1)(激发波长471 nm,发射波长517 nm);图8. 草甘膦荧光分子印迹纳米粒子(mip1)对不同浓度的草甘膦的荧光响应曲线;图9. 不同试纸与茚三酮的反应结果(a)未处理试纸(b)修饰氨基后的试纸(c)修饰氨基后修饰醛基的试纸;图10. mip1-@pc(左)和nip1-@pc(右)在365 nm紫外光线照射下的荧光图;图11. 草甘膦荧光分子印迹试纸对不同浓度的草甘膦的响应分析(mip1@pc、nip1@pc);
图12. 草甘膦荧光分子印迹纳米粒子对不同农药的响应分析(mip1@pc、nip1@pc);图13. 草甘膦荧光分子印迹纳米粒子(mip1@pc)对不同量的草甘膦的荧光响应线性范围。
[0024]
具体实施方式
[0025]
下面结合具体实施例和附图对本发明进一步进行说明。
[0026]
实施例中所述实验方法,如无特殊说明,均为常规方法;所述试剂和材料,如无特殊说明,均可从商业途径获得。
[0027]
实施例1制备荧光单体:将异硫氰酸荧光素(fitc, 1.6 mg,4 μmol)溶于0.8 ml 无水乙醇中,加入3-氨丙基三乙氧基硅烷(aptes, 0.936 μl, 4 μmol)超声混匀,锡纸包裹,放入摇床混合5 h可得aptes-fitc 荧光单体。在相同情况下配制 aaptms-fitc 和 aaaptms-fitc荧光单体,其中n-[3-(三甲氧基硅基)丙基]乙二胺(aaptms, 4 μmol),3-[2-(2-氨基乙基氨基)乙基氨基]丙基-三甲氧基硅烷(aaaptms, 4 μmol)。
[0028]
实施例2制备分子印迹聚合物,具体合成路线如图2所示,制备方法如下:在圆底烧瓶中加入纯水/无水乙醇(20 ml/12 ml)的共溶剂搅拌 5 min;使用3-氨丙基三乙氧基硅烷(aptes,140 μl,0.6 mmol)作为功能单体和模板草甘膦(16.9 mg,0.1 mmol)搅拌 10 min;后加入荧光功能单体fitc
ꢀ‑ꢀ
aptes(0.4 ml,2 μmol)搅拌10 min;四乙氧基硅酸酯(teos,670 μl,3 mmol)搅拌48 h。离心后经水、乙醇、磷酸盐洗涤后去除模板草甘膦,得到草甘膦分子印迹聚合物mip1,产率为16.4%。相同的方法制备不含模板草甘膦的非分子印迹聚合物nip1,产率为29.69%。
[0029]
上述聚合物均进行了扫描电镜表征,如图3所示。
[0030]
实施例3同实施例2,区别在于功能单体3-氨丙基三乙氧基硅烷(aptes)换为n-[3-(三甲氧基硅基)丙基]乙二胺(aaptms),制备所得的草甘膦分子印迹聚合物记为mip2,非分子印迹聚合物记为nip2,产率分别为48.72%、39.13%。
[0031]
实施例4同实施例2,区别在于功能单体3-氨丙基三乙氧基硅烷(aptes)换为3-[2-(2-氨基乙基氨基)乙基氨基]丙基-三甲氧基硅烷(aaaptms),制备所得的草甘膦分子印迹聚合物记为mip3,非分子印迹聚合物记为nip3,产率分别为34.09%、26.02%。
[0032]
实施例5对实施例2制备的草甘膦荧光分子印迹聚合物进行选择性测试将实施例2中制备的聚合物制备成一系列浓度梯度的mip1和nip1溶液。以纯水为溶剂配置0.01 mg/ml、0.05 mg/ml、0.1 mg/ml、0.5 mg/ml浓度的mip1和nip1溶液,振荡超声使其混合均匀。样液混合均匀后取2 ml 0.01 mg/ml mip1、nip1溶液分别放入两个比色皿中,置于磁力搅拌器上搅拌15 min,将比色皿置于荧光分光光度计中进行检测(激发波长
471 nm,发射波长517 nm)记作空白值f0,向比色皿中加入10 μl 5 μmol/l草甘膦溶液,置于磁力搅拌器上搅拌15 min,测量其荧光响应强度,记作f,计算获得荧光响应变化值((f
0-f)/f0),使用相同的方法测得纳米粒子浓度为0.05 mg/ml、0.1 mg/ml、0.5 mg/ml荧光响应变化值。
[0033]
如图4所示0.01 mg/ml、0.05 mg/ml、0.1 mg/ml、0.5 mg/ml浓度的mip1的荧光响应变化率均高于它们的对应荧光纳米粒子0.01 mg/ml、0.05 mg/ml、0.1 mg/ml、0.5 mg/ml浓度的nip1,0.05 mg/ml和0.1 mg/ml的 mip1荧光响应变化率分别显著高于0.05 mg/ml和0.1 mg/ml nip1,表明该分子印迹荧光纳米粒子对草甘膦具有良好的响应选择性。印迹因子指目标物被mip吸附的量或者引起的信号变化与被nip 吸附的量或者引起的信号变化的比值,可用于表示选择性的高低,印迹因子越高说明选择性越高,由图可知印迹因子分别为4.7、7.6、6.0、1.5,由此可见该荧光分子印迹纳米粒子对草甘膦的选择性较高。
[0034]
实施例6对实施例3制备的草甘膦荧光分子印迹聚合物进行选择性测试将实施例3中制备的聚合物制备成一系列浓度梯度的mip2和nip2溶液。以纯水为溶剂配置0.01 mg/ml、0.05 mg/ml、0.1 mg/ml、0.5 mg/ml浓度的mip2和nip2溶液,振荡超声使其混合均匀。样液混合均匀后取2 ml 0.01 mg/ml mip2、nip2溶液分别放入两个比色皿中,置于磁力搅拌器上搅拌15 min,将比色皿置于荧光分光光度计中进行检测(激发波长471 nm,发射波长517 nm)记作空白值f0,向比色皿中加入10 μl 5 μmol/l草甘膦溶液,置于磁力搅拌器上搅拌15 min,测量其荧光响应强度,记作f,计算获得荧光响应变化值((f
0-f)/f0),使用相同的方法测得纳米粒子浓度为0.05 mg/ml、0.1 mg/ml、0.5 mg/ml荧光响应变化值。
[0035]
如图5所示0.01 mg/ml、0.05 mg/ml、0.1 mg/ml、0.5 mg/ml浓度的mip2的荧光响应变化率均高于它们的对应荧光纳米粒子0.01 mg/ml、0.05 mg/ml、0.1 mg/ml、0.5 mg/ml浓度的nip2,0.05 mg/ml和0.1 mg/ml的 mip2荧光响应变化率分别显著高于0.05 mg/ml和0.1 mg/ml nip2,表明该分子印迹荧光纳米粒子对草甘膦具有良好的响应选择性。印迹因子指目标物被mip吸附的量或者引起的信号变化与被nip 吸附的量或者引起的信号变化的比值,可用于表示选择性的高低,印迹因子越高说明选择性越高,由图可知印迹因子分别为1.1、1.0、3.5、1.3,由此可见该荧光分子印迹纳米粒子对草甘膦的选择性较高。
[0036]
实施例7对实施例4制备的草甘膦荧光分子印迹聚合物进行选择性测试将实施例4中制备的聚合物制备成一系列浓度梯度的mip3和nip3溶液。以纯水为溶剂配置0.01 mg/ml、0.05 mg/ml、0.1 mg/ml、0.5 mg/ml浓度的mip3和nip3溶液,振荡超声使其混合均匀。样液混合均匀后取2 ml 0.01 mg/ml mip3、nip3溶液分别放入两个比色皿中,置于磁力搅拌器上搅拌15 min,将比色皿置于荧光分光光度计中进行检测(激发波长471 nm,发射波长517 nm)记作空白值f0,向比色皿中加入10μl 5μmol/l草甘膦溶液,置于磁力搅拌器上搅拌15 min,测量其荧光响应强度,记作f,计算获得荧光响应变化值((f
0-f)/f0),使用相同的方法测得纳米粒子浓度为0.05 mg/ml、0.1 mg/ml、0.5 mg/ml荧光响应变化值。
[0037]
如图6所示0.01 mg/ml、0.05 mg/ml、0.1 mg/ml、0.5 mg/ml浓度的mip3的荧光响
mip1-@pc 的荧光固载强度高于 nip1-@pc,推测和聚合液配方的成分相关,间接表明模板分子的加入会影响聚合反应的效率。利用草甘膦荧光分子印迹进行检测时,向试纸上滴加草甘膦样品,其物质扩散会以咖啡环的形式进行扩散如图1所示,会形成环状斑现象,使用倒置荧光显微镜对试纸的荧光信号测试时,测试位置的选择利用了咖啡环效应即选择1区和2区之间的环状位置测试。
[0048]
实施例12对实施例11制备的草甘膦分子印迹试纸进行选择性测试首先使用倒置荧光显微镜在529 nm处测得试纸的初始荧光值f0,配制草甘膦溶液,依次向试纸的中心位置上滴加草甘膦溶液(滴加方法如图1所示,产生咖啡圈效应,将样品滴加到试纸中心位置,液体会扩散,其中的草甘膦会被吸附在中心点,其他干扰物会随着咖啡圈效应扩散到试纸边缘)使试纸上的草甘膦含量分别达到0.1 μmol、0.5 μmol、1 μmol、5 μmol、8 μmol、10 μmol,使用倒置荧光显微镜测得荧光强度f分别记为f0.1、f0.5、f1、f5、f8、f10,计算荧光变化f
0-f,同理可得nip1-@pc的荧光变化,如图11所示可得印迹因子分别为1.4、1.4、1.4、1.2、1.3、1.6,由此可知mip-@pc具有良好的选择性。
[0049]
实施例13对实施例11制备的草甘膦分子印迹试纸进行特异性测试。
[0050]
草甘膦、吡虫啉、2,4-d、毒死蜱分别配成1 mmol/l的溶液,用荧光倒置显微镜测量 mip1-@pc 荧光值,记为 f0。加入 20 μl 1 mmol/l 的吡虫啉,记为 f,计算荧光变化率;用同样的方法测得mip1-@pc的荧光变化率。如图12所示,mip-@pc对草甘膦的荧光响应变化值高于对其他农药的响应变化值,mip-@pc对草甘膦具有良好的特异性。
[0051]
实施例14对实施例11制备的草甘膦分子印迹试纸的标准曲线的建立。
[0052]
mip-@pc使用倒置荧光显微镜在529 nm处测得f0,向试纸上分别滴加不同浓度的草甘膦使试纸上草甘膦的量分别为0.1、0.5、1、2、5、8、10 μmol,试纸干燥后测得f,计算荧光变化值,如图13可以看出,在mip1-@pc 的线性范围为 0.5-10 μmol,最低检测限为 0.29 μmol。
[0053]
实施例15用于实际样品(自来水和大豆样品)检测。
[0054]
将大豆样品按四分法缩分出约1 kg,全部粉碎并通过20目筛,混匀,均分成两份试样,装入洁净的容器内,密封,标记,常温保持。称取10 g试样于150 ml离心瓶中加入水至含水量达50 ml,同时加入一定量的草甘膦样品,混匀后浸泡0.5小时,高速均质5 min,于3500 r/min离心10 min,取上清液过滤两次,即得草甘膦大豆滤液。
[0055]
向试纸上滴加草甘膦大豆滤液,使试纸上草甘膦的量分别为0.5、1、2 μmol。使用倒置荧光显微镜在529 nm处测得试纸的初始荧光值f0,再测得添加草甘膦后的试纸荧光强度f,通过计算(f
0-f)/f0得到荧光变化率,再依据实施例14制作标准曲线的步骤进行本法的加标回收率的测定,并计算加标回收率。用自来水作为样品,配制自来水草甘膦溶液,测试方法同上。回收率=(样品回收检测值-空白回收检测值)/标准品检测值*100%,计算回收率,结果如表1所示,待测样品的加标回收率在91.74-117.42%之间,证明了该方法的准确性和可靠性。
[0056]
表1加标回收率的测定
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。