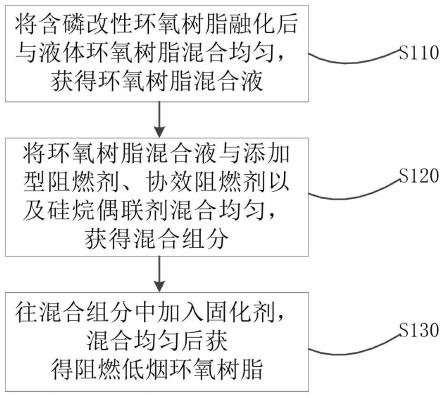
1.本发明涉及高分子聚合物技术领域,具体而言,涉及一种亚光化耐冲击高抗张强度的树脂组合物及其制备方法。
背景技术:
2.abs树脂组合物(丙烯腈-丁二烯-苯乙烯共聚物)是以橡胶状接枝共聚物分散于苯乙烯系共聚物中所制成的一种抗冲击树脂组合物。当用于家电制品的外壳、皮箱和汽车仪表板的外皮等,经常要求既满足耐冲击、平衡的拉伸性能同时又需要具备亚光化的表面特性,以适应人类对于制品外观的个性化追求。
3.一般而言,通常耐冲击愈高的abs树脂,光泽度愈低,换句话说,abs树脂的橡胶含量愈高、橡胶粒子愈大光泽度就愈低,ep 201,099提供了一种两步法聚合工艺制备低光泽abs(丙烯腈-苯乙烯-丁二烯)树脂的方法;ep 129,796中描述了将苯乙烯及丙烯腈接枝于聚丁二烯橡胶上制备abs接枝共聚物的途径;us 3,928,494、de 2,057,935等专利进一步描述了高抗冲abs的制备工艺。但是提高胶量abs树脂的抗张强度劣化、增大粒径所需要的合成技术相对又比较复杂,这两种方法尽管对光泽的降低有作用,但要获得在不同领域、场合下实际所需光泽度却有很大难度。
4.专利jp 58-93711揭示了一种低光泽abs热塑性树脂的制备过程,其方法是将聚苯乙烯混入abs胶乳中,利用其相容性差及两种材料收缩率的差异,降低光泽。但是,这种方法在降低树脂表面光泽上效果并不显著,并因此丧失了产品的冲击强度。
5.用一种特定的乳液聚合方法可以制备低光泽abs树脂,其特点为在聚合过程中加入羧酸类单体,利用缩聚反应生成的聚合物颗粒降低表面光泽。但由于缩聚反应低的产率,所以其对光泽的降低贡献微乎其微,更何况其生产重现性不好。
6.专利kp 93-6912用悬浮或本体-悬浮两步法聚合工艺,制造低光泽abs树脂。这种abs随着注射条件的不同又出现一些诸如严重收缩、光泽分布不均、硬度降低等问题,限制了其应用范围。
7.专利wo 99/03904中所述的低光泽abs树脂,是在abs中加入聚合物光泽改性剂,达到降低表面光泽的目的。这种聚合物光泽改性剂是在abs胶乳凝聚阶段加入不超过25%的pbd或sbr或nbr胶乳共凝制成。它的缺陷在于所制备的abs组合物的光泽虽然得到了有效的降低,但并未照顾到冲击强度、拉伸强度综合性能的平衡,实际应用时,特别在光泽需要频繁调整时又显得很不方便。
8.此外,在树脂组合物中添加有机或无机消光剂,如凝胶化的聚合物、超高分子量聚合物、氧化锌、二氧化钛、二氧化硅等物质亦可起到消光效果,但其往往牺牲了聚合物材料宝贵的韧性;另一种常用的方法是改进加工技术,常见的比如表面皮纹化等。这两种方法均对材料的某些性能如机械性能、染色性能等造成负面影响,并提高了最终产品的成本。
9.因此,如何方便的制备一种同时具有耐冲击性、高抗张强度和制品表面亚光化设计的树脂组合物(特别是abs树脂),一直是业界人士长期以来研究的课题。
技术实现要素:
10.本发明第一方面涉及一种树脂组合物,以重量份计,包括20~48份a、50~80份b、1.5~7份c以及0.2~1.5份d,其中:
11.a为由40~70份二烯系橡胶和30~60份的第一单体混合物聚合得到的改性树脂,且所述二烯系橡胶的平均粒径为0.15um~0.7um;
12.b为由第二单体混合物聚合而得到的树脂;
13.所述第一单体混合物和所述第二单体混合物独立地包含60~90份的苯乙烯系单体和10~40份的丙烯腈系单体;
14.c为丁腈胶乳制备得到的聚合物粉状体,其丁腈胶乳凝胶含量为50%~95%,门尼黏度为65~130ml;
15.d为助剂。
16.根据本发明的再一方面,还涉及如上所述树脂组合物的制备方法,包括将各组分共混成型。
17.本发明利用两种材料之间收缩率、折光性能等方面存在的差异,经共混等手段制备了一种光泽可方便调整、又同时兼有好的冲击强度、抗张强度的树脂组合物。
具体实施方式
18.现将详细地提供本发明实施方式的参考,其一个或多个实例描述于下文。提供每一实例作为解释而非限制本发明。实际上,对本领域技术人员而言,显而易见的是,可以对本发明进行多种修改和变化而不背离本发明的范围或精神。例如,作为一个实施方式的部分而说明或描述的特征可以用于另一实施方式中,来产生更进一步的实施方式。
19.除非另有说明,用于披露本发明的所有术语(包括技术和科学术语)的意义与本发明所属领域普通技术人员所通常理解的相同。通过进一步的指导,随后的定义用于更好地理解本发明的教导。本文中在本发明的说明书中所使用的术语只是为了描述具体的实施例的目的,不是旨在于限制本发明。
20.本文所使用的术语“和/或”、“或/和”、“及/或”的选择范围包括两个或两个以上相关所列项目中任一个项目,也包括相关所列项目的任意的和所有的组合,所述任意的和所有的组合包括任意的两个相关所列项目、任意的更多个相关所列项目、或者全部相关所列项目的组合。需要说明的是,当用至少两个选自“和/或”、“或/和”、“及/或”的连词组合连接至少三个项目时,应当理解,在本技术中,该技术方案毫无疑问地包括均用“逻辑与”连接的技术方案,还毫无疑问地包括均用“逻辑或”连接的技术方案。比如,“a及/或b”包括a、b和a b三种并列方案。又比如,“a,及/或,b,及/或,c,及/或,d”的技术方案,包括a、b、c、d中任一项(也即均用“逻辑或”连接的技术方案),也包括a、b、c、d的任意的和所有的组合,也即包括a、b、c、d中任两项或任三项的组合,还包括a、b、c、d的四项组合(也即均用“逻辑与”连接的技术方案)。
21.本发明中所使用的术语“含有”、“包含”和“包括”是同义词,其是包容性或开放式的,不排除额外的、未被引述的成员、元素或方法步骤。
22.本发明中用端点表示的数值范围包括该范围内所包含的所有数值及分数,以及所引述的端点。
23.本发明中涉及浓度数值,其含义包括在一定范围内的波动。比如,可以在相应的精度范围内波动。比如2%,可以允许
±
0.1%范围内波动。对于数值较大或无需过于精细控制的数值,还允许其含义包括更大波动。比如100mm,可以允许
±
1%、
±
2%、
±
5%等范围内的波动。涉及分子量,允许其含义包括
±
10%的波动。
24.本发明中,涉及“多个”、“多种”等描述,如无特别限定,指在数量上指大于等于2。
25.本发明中,所有的百分数(%)数除注明外,均为重量百分数。
26.本发明中,所有的份数除注明外,均为重量份数。
27.本发明中,以开放式描述的技术特征中,包括所列举特征组成的封闭式技术方案,也包括包含所列举特征的开放式技术方案。
28.本发明中,“优选”、“更好”、“更佳”、“为宜”仅为描述效果更好的实施方式或实施例,应当理解,并不构成对本发明保护范围的限制。
29.在本发明提及的所有文献都在本技术中引用作为参考,就如同每一篇文献被单独引用作为参考那样。除非和本技术的发明目的和/或技术方案相冲突,否则,本发明涉及的引用文献以全部内容、全部目的被引用。本发明中涉及引用文献时,相关技术特征、术语、名词、短语等在引用文献中的定义也一并被引用。本发明中涉及引用文献时,被引用的相关技术特征的举例、优选方式也可作为参考纳入本技术中,但以能够实施本发明为限。应当理解,当引用内容与本技术中的描述相冲突时,以本技术为准或者适应性地根据本技术的描述进行修正。
30.本发明第一方面涉及一种树脂组合物,以重量份计,包括20~48份a、50~80份b、1.5~7份c以及0.2~1.5份d,其中:
31.a为由40~70份二烯系橡胶和30~60份的第一单体混合物聚合得到的改性树脂,且所述二烯系橡胶的平均粒径为0.15um~0.7um;
32.b为由第二单体混合物聚合而得到的树脂;
33.所述第一单体混合物和所述第二单体混合物独立地包含60~90份的苯乙烯系单体和10~40份的丙烯腈系单体;
34.c为丁腈胶乳制备得到的聚合物粉状体,其丁腈胶乳凝胶含量为50%~95%,门尼黏度为65~130ml;
35.d为助剂。
36.本发明所制备得到的树脂组合物具有耐冲击、高抗张强度以及光泽可调的特性。
37.在一些实施方式中,所述二烯系橡胶是指其单体包含二烯结构的化合物,优选为至少包含丁二烯,还可以包含苯乙烯、丙烯腈、甲基丙烯酸甲酯等单体。在一些实施方式中,所述二烯系橡胶包括丁二烯-苯乙烯共聚物、丁二烯-丙烯腈共聚物、丁二烯-甲基丙烯酸甲酯共聚物和聚丁二烯中的一种或多种,优选为聚丁二烯。
38.本发明所采用的二烯系橡胶可以由单体直接聚合成粒径为0.15um~0.7um的胶乳,也可先合成0.05μm~0.18μm的小粒径胶乳后,再用化学的或物理的方法附聚成所需大小的粒子。
39.在一些实施方式中,所述苯乙烯系单体包括苯乙烯、α-甲基苯乙烯、α-氯苯乙烯或p-甲基苯乙烯中的一种或多种,优选苯乙烯。
40.在一些实施方式中,所述丙烯腈系单体包括丙烯腈和/或α-甲基丙烯腈单体,优选
丙烯腈。
41.在上述组分中,a为20~48份,还可以选择24、28、32、36、40、44份。当其含量低于20份时,树脂无法得到所需要的韧性与光泽的统一;而大于50份时,树脂又无法得到所需要的抗张强度。
42.在优选的实施方式中,a的接枝率在40%~70%之间,例如50%、60%;接枝上的硬质聚合物的数均分子量优选在50000~200000之间,例如70000、100000、130000、150000、170000。
43.a的平均粒径为0.15um~0.7um,例如0.2um、0.3um、0.4um、0.5um、0.5um。a优选为由60~70份二烯系橡胶和30~40份的第一单体混合物采用乳液聚合而得到的改性树脂。
44.在上述组分中,b为50~80份,还可以选择55、60、65、70、75份。b小于50份时组合物的抗张强度不足,而大于80份时组合物的韧性又不够。
45.在一些实施方式中,b为苯乙烯-丙烯腈共聚物。
46.在一些实施方式中,所述第二单体混合物中苯乙烯为60~80份,丙烯腈为20~40份。
47.在一些实施方式中,c中丁腈胶乳凝胶含量为50%~70%,门尼黏度为65~90ml(简称c2,或半交联型);或者c中丁腈胶乳凝胶含量为70%~95%,门尼黏度为90~130ml(简称c1,或交联型)。
48.组合物中的半交联结构(c2)组分橡胶状聚合物的引入可提高组合物的冲击强度,改进组合物的加工性能,降低树脂的表面光泽,其效果优于交联结构的(c1);当(c)的用量在1.5~7.0份时,可获得本发明所说之较佳效果。不使用本发明(c)时无法得到亚光化的树脂组合物。
49.在一些实施方式中,所述助剂包括热稳剂、润滑剂、抗氧剂、填充剂、颜料、抗静电剂和分散剂中的一种或多种,且不包括消光剂。
50.本发明的第二方面涉及如上所述树脂组合物的制备方法,包括将各组分共混成型。
51.其共混可以通过采用常用的方法,例如使用螺带式掺混机、亨舍尔混合机、班伯里混合机、转鼓、单螺杆挤压机、捏和机、间歇混合机、双螺杆挤压机、共混机、多螺杆挤压机等来进行。
52.本发明的树脂组合物,可以制造各种成型品、片材、膜。作为此时的成型方法,可以使用对于树脂组合物通常使用的各种成型方法,例如可以采用注射成型、挤出成型、加压成型、吹塑成型、压延成型、流延成型等任意的成型法。此外,还可以采用膜、片材的成型中通常的t模头法、压延法、吹胀法、带法等。
53.此外,共混时优选进行加热,加热温度通常在170℃~300℃,优选220~270℃的范围选择。
54.本发明的聚合物a可通过接枝聚合反应制造得到。通常采用乳液接枝的技术,将苯乙烯系、丙烯腈系单体混合物利用化学性的结合或接枝将至少一种聚合物连接到橡胶相分子链上,依单体与丁二烯系橡胶的比例及聚合条件,可得到所希望的接枝程度的聚合物,通常接枝聚合反应中的聚合条件、橡胶状聚合物的化学性质、粒子大小、单体加入的速度、方式、链转移剂、乳化剂的用量及种类等因素,都会影响其接枝的程度。
55.前述聚合反应所使用的引发剂,可以是水溶性的,也可以是油溶性的,用量优选0.1%~1.5%(以聚合单体100重量份计),为确保聚合反应的顺利进行,引发剂以连续或增量法加入效果最好。接枝共聚物的分子量大小由接枝反应的温度加以控制,和/或配合相当少量常用的分子量调节剂,如:正—十二烷基硫醇、叔—十二烷基硫醇等。
56.反应中接枝橡胶含量的控制可以利用单体混合物的连续或增量的加入,并最好同时连续或增量的加入引发剂、调节剂,以确保最终接枝产品综合性能的平衡。
57.聚合反应通常在搅拌釜中进行,常压或略带压的装置均可,要达到96%以上的单体转化率,聚合时间5~8小时就足够了。在一些实施方式中,a采用乳液聚合制备得到。
58.经上述乳液聚合可以制得接枝共聚物a的乳液,a的乳液中加入适当的凝聚剂进行凝聚。一般凝聚剂可选酸类、碱土金属盐等。凝结完成的浆液经离心脱水除去大部分水分,再经干燥处理,制得粉状接枝共聚物a。
59.在一些具体的实施方式中,a的制备过程是将配方量的单体和化学品,依次加入聚合反应器中,升温到70℃~75℃,并恒温常压反应5~8小时,分析转化率》96.0%时,然后夹套内通冷水把聚合液降温到40℃以下,再用8~10%的硫酸镁溶液凝聚,离心、脱水、鼓风干燥获得接枝共聚物a。
60.聚合物b聚合的方法可采用溶液聚合、本体聚合、悬浮聚合或乳液聚合制备得到。b的制备方法是业界所熟知的。
61.橡胶弹性体c可以用喷雾干燥、化学凝聚、活性隔离的方法生产的橡胶粉,也可使用块状胶经冷冻粉碎制得的橡胶粉。
62.橡胶弹性体c优选采用活性隔离法制备得到,示例性的制备过程如下:
63.用隔离剂超细碳酸钙对丁腈胶乳进行隔离处理,隔离剂用量相当于胶乳(干基)的2%~3%,然后分3批(1/3,1/3,1/3)连续加入8%~10%的盐溶液到丁腈胶乳中进行凝聚,凝聚剂用量相当于胶乳(干基)的4.0%~5.0%。凝聚优选使用500l不朽钢凝聚釜,透平浆式三叶搅拌,搅拌转速优选为300rpm~600rpm。通过聚合釜夹套控制凝聚体系温度为45℃~60℃,保持凝聚时间1.5~2.0小时,常压下完成凝聚,然后升温到75℃~80℃熟化固化30~40分钟,经过滤、洗涤、脱水、旋风干燥制得粉末橡胶弹性体c。
64.下面将结合实施例对本发明的实施方案进行详细描述。应理解,这些实施例仅用于说明本发明而不用于限制本发明的范围。下列实施例中未注明具体条件的实验方法,优先参考本发明中给出的指引,还可以按照本领域的实验手册或常规条件,还可以参考本领域已知的其它实验方法,或者按照制造厂商所建议的条件。
65.下述的具体实施例中,涉及原料组分的量度参数,如无特别说明,可能存在称量精度范围内的细微偏差。涉及温度和时间参数,允许仪器测试精度或操作精度导致的可接受的偏差。
66.下述实施例、比较例所采用的分析测试方法和标准如下:
67.1.abs接枝橡胶含量分析:
68.称取试样1.5克(准确至0.0002克)置于凯式烧瓶内,沿瓶颈加入100ml丙酮,将冷凝管装在烧瓶上,在65℃恒温水浴上回流2-3小时。在此期间勿使水浴温度高于65℃,否则易发生暴沸现象。
69.停止加热后取下烧瓶,冷却至室温。将烧瓶内的内容物全部转入已称量过的聚乙
烯离心管内,放置于离心机中,在20000rpm下离心分离30分钟,然后将上层清液倾入100ml三角瓶内。用少量丙酮洗涤离心管内不溶物(用玻璃棒搅拌)后再离心分离一次,除去上层清液。将离心管放入真空烘箱内,于65℃下真空干燥至恒重后称量(g1)。
70.a.结果计算
71.接枝橡胶含量x(wt%)按下式计算:
[0072][0073]
式中,g1=橡胶重量,克;
[0074]
g=试样重量,克。
[0075]
试样的测定应平行进行两次,结果计算至小数第二位,经平均和数字修约后,分析报告取小数后一位。
[0076]
另外,通过本试验测定,也可按下式求出接枝效率:
[0077][0078]
式中,san总量=试样重量
×
配方中苯乙烯和丙烯腈所占的百分含量。
[0079]
1.2abs中的san分子量及其分布分析
[0080]
称取试样1.5克(准确至0.0002克)置于凯式烧瓶内,沿瓶颈加入100ml丙酮,将冷凝管装在烧瓶上,在65℃恒温水浴上回流2-3小时。在此期间勿使水浴温度高于65℃,否则易发生暴沸现象。
[0081]
停止加热后取下烧瓶,冷却至室温。将烧瓶内的内容物全部转入已称量过的聚乙烯离心管内,放置于离心机中,在20000rpm下离心分离30分钟,然后将上层清液倾入100ml三角瓶内。用少量丙酮洗涤离心管内不溶物(用玻璃棒搅拌)后再离心分离一次,除去上层清液,取其清液进行干燥而后制得san样或直接用清液做为样品。准备好的试样用四氢呋喃(thf)作为溶剂溶解,然后用gpc法(与体积排斥色谱是一回事)进行分析。
[0082]
a.试验仪器
[0083]
美国沃特斯150-c gpc/alc或其它相同类型的凝胶渗透色谱(gpc)仪;
[0084]
真空过滤器(带小型无油真空泵);
[0085]
数据处理机(使用沃特斯的色谱处理系统)。
[0086]
b.溶剂
[0087]
专业gpc分析所用的试剂在常温下一般都是thf,它的特点是对大部分聚合物溶解力强、折光指数低,而且与水互溶的特性使得直接溶解胶乳进行分析成为可能。另外,thf在使用之前要进行脱气处理,以保证系统的稳定性等。
[0088]
c.测定分析步骤
[0089]
(1)按仪器中分离柱的要求或自己试验优选的浓度大小溶解样品;
[0090]
(2)启动仪器,并用过滤好的新鲜溶剂置换稳定系统。通常,此时的流速设定为0.1~0.2ml/min为好;
[0091]
(3)过滤溶解好的样品,用专用样品瓶把过滤后的样品放入仪器注射进样室恒温,设置仪器工作参数。主要是液体流动相的流速和柱温,流速为1.0ml/min,柱温为30℃;
[0092]
(4)进样,采集相关信号;
[0093]
(5)处理采集到的数据,得出各种平均分子量,并打印出分子量分布谱图。
[0094]
1.3接枝胶乳总固物的测定
[0095]
在已知重量的铝盘上称取1.5g试样(准确至0.0002g),滴加约3g乙醇于试样中使其凝聚。放入红外线干燥器中干燥到变成土黄色(约10分钟),冷却后称量。(当取样量多或总固物含量高时,乙醇量要加到完全凝固为止。
[0096]
计算:
[0097]
总固物含量x(重量%)按下式计算:
[0098][0099]
式中:g2——总固体物重 铝盘重,克
[0100]
g1—铝盘重,克
[0101]
g—试样重,克
[0102]
1.4聚合物的丙烯腈含量的分析,可通过经典的化学定氮法进行较好的定量分析(可参照sh/t 1157-1997,等效于iso 1656:1988)
[0103]
1.5物理机械性能测试相关标准
[0104][0105]
光泽度:参照astm d523,jis k7105标准
[0106]
制备实施例聚合物的制备方法
[0107]
聚合物a、c的制备过程及物理特性:
[0108]
1.先按以下配方制备橡胶接枝共聚物(a):
[0109]
[0110][0111]
聚合物a的制备过程:
[0112]
将上述配方量的化学物质,依次加入3.0l聚合反应器中,在1.5小时内通过夹套水浴,使聚合液升温到70-75℃,恒温常压反应5.0-6.0小时,分析转化率》96.0%时,然后夹套通冷水把物料降温到40℃以下,再用8-10%的硫酸镁溶液凝聚,离心、脱水、干燥获得聚合物a。
[0113]
聚合物a的物理特性如下:
[0114]
橡胶含量65%(以聚丁二烯计,配方计算结果),重量平均粒径为0.485μm(粒度分析仪直接测定),接枝橡胶含量85.6%(化学分离法分析,通用的分析方法),苯乙烯-丙烯腈共聚物的分子量为69,000(凝胶色谱法,以聚苯乙烯为基准)。
[0115]
一般的,本发明所要求的(a)的接枝率应在40~70%之间,接枝上的硬质聚合物的数均分子量在50,000~200,000之间。
[0116]
2.采用1994年03月16日公开的专利申请cn1083827a所提供的方法制成橡胶弹性体(c)*;
[0117]
{(c)*因所使用胶乳凝胶及门尼黏度不同可分为交联型(c1:凝胶70~95%,门尼黏度90~130ml)、半交联型(c2:凝胶50~70,门尼黏度65~90ml)}
[0118]
橡胶弹性体(c)制备过程如下:
[0119]
用隔离剂超细碳酸钙对丁腈胶乳进行隔离处理,隔离剂用量相当于胶乳(干基)的2-3%,然后分3批(1/3,1/3,1/3)连续加入8-10%的盐溶液到丁腈胶乳中进行凝聚,凝聚剂用量相当于胶乳(干基)的4.0-5.0%,凝聚使用500l不朽钢凝聚釜,透平浆式三叶搅拌,搅拌转速400rpm。通过聚合釜夹套控制凝聚体系温度为45℃~60℃,保持凝聚时间1.5~2.0小时,常压下完成凝聚,然后升温到80℃~85℃熟化固化30分钟,经过滤、洗涤、脱水、旋风干燥制得粉末丁腈—橡胶弹性体(c)。
[0120]
橡胶弹性体(c)性状:
[0121]
白色粉末,灰份小于4%,丙烯腈含量26.8-29.0%,粒度90%在40-60目之间,依据凝胶含量差异制备两种门尼粘度不同的胶粉c1(交联型)和c2(半交联型)。
[0122]
以下实施例、比较例均在5l高速混合器中,常温混合3分钟(800rpm/60s 3000rpm/60s 1500rpm/60s),然后混合料经直径45mm(长径比42)双螺杆挤出机,在190℃~240℃的温度下挤出造粒制得。
[0123]
所得的样品在80℃鼓风干燥箱内烘干2~4小时,最后按照gb标准进行注射,注射工艺参数如下:
[0124]
机筒温度:190℃~240℃
[0125]
注射压力:65mpa~110mpa
[0126]
模具温度:40℃~80℃
[0127]
注射速度:尽量缓慢。
[0128]
实施例-1
[0129]
将465克a,1535克b(悬浮法san,悬浮法生产的苯乙烯-丙烯腈共聚物),6克硬脂酸镁,6克ebs(乙撑双硬脂酰亚胺),60克c2共混后挤出造粒,组合物性能为:冲击强度351.2j/m(焦耳/米,下同),抗张强度38.5mpa(兆帕,下同),mi(熔体指数)1.8g/10min(克/10分钟),表面光泽(600度)76。
[0130]
而不加入c2时表面光泽(600度)是96,其他性能依次是316、39、1.9。
[0131]
实施例-2
[0132]
将430克a,1570克b(同实施例-1),6克硬脂酸镁,6克ebs,100克c2共混后挤出造粒,组合物性能为:冲击强度335.1j/m,抗张强度38.8mpa,mi1.67g/10min,表面光泽(600)72。
[0133]
而不加入c2时表面光泽(600度)是96.5,其他性能依次是302、39、2.2。
[0134]
实施例-3
[0135]
将630克a,1370克b(本体san,苯乙烯-丙烯腈共聚物),6克硬脂酸镁,6克ebs,60克c2共混后挤出造粒,组合物性能为:冲击强度417.8j/m,抗张强度37.9mpa,mi1.3g/10min,表面光泽(600)73。
[0136]
而不加入c2时表面光泽(600度)是94,其他性能依次是336、38、1.4。
[0137]
实施例-4
[0138]
将630克份a,1370克b(本体san),6克硬脂酸镁,6克ebs,80克c2共混后挤出造粒,组合物性能为:冲击强度436.8j/m,抗张强度37.5mpa,mi1.2g/10min,表面光泽(600)68。
[0139]
而不加入c2时表面光泽(600度)是93,其他性能依次是336、38、1.4。
[0140]
实施例-5
[0141]
将630克a,1370克b(本体san),6克硬脂酸镁,6克ebs,100克c2共混后挤出造粒,组合物性能为:冲击强度467.2j/m,抗张强度37.5mpa,mi1.1g/10min,表面光泽(600)63。
[0142]
而不加入c2时表面光泽(600度)是93,其他性能依次是336、38、1.4。
[0143]
实施例-6
[0144]
将460克a,1540克b(本体san),6克硬脂酸镁,6克ebs,60克c1共混后挤出造粒,组合物性能为:冲击强度317.8j/m,抗张强度41.0mpa,mi1.9g/10min,表面光泽(600)78。
[0145]
而不加入c1时表面光泽(600度)是96,其他性能依次是306、39、1.8。
[0146]
实施例-7
[0147]
将440克a,1560克b(本体san),6克硬脂酸镁,6克ebs,80克c1共混后挤出造粒,组合物性能为:冲击强度298.5j/m,抗张强度42.1mpa,mi2.0g/10min,表面光泽(600)75。
[0148]
而不加入c1时表面光泽(600度)是96.5,其他性能依次是298、42、2.0。
[0149]
实施例-8
[0150]
将420克a,1580克b(本体san),8克硬脂酸镁,8克ebs,100克c1共混后挤出造粒,组合物性能为:冲击强度269.5j/m,抗张强度41.7mpa,mi2.0g/10min,表面光泽(600)73。
[0151]
而不加入c1时表面光泽(600度)是97,其他性能依次是256、43、2.1。
[0152]
实施例-9
[0153]
将360克a,1640克b(悬浮法san),6克硬脂酸镁,6克ebs,100克c1共混后挤出造粒,组合物性能为:冲击强度232.7j/m,抗张强度39.7mpa,mi2.2g/10min,表面光泽(600)72。
[0154]
而不加入c1时表面光泽(600度)是98,其他性能依次是213、46、2.3。
[0155]
比较例
[0156]
比较例-1
[0157]
将280克a,1720克b(悬浮法san),6克硬脂酸镁,6克ebs,100克c1共混后挤出造粒,组合物性能为:冲击强度157.7j/m,抗张强度43.1mpa,mi2.4g/10min,表面光泽(600)79。
[0158]
比较例-1说明:不使用本发明规定的a、b用量范围,使用c1虽可一定程度调整产品的光泽,但冲击强度并不理想。
[0159]
比较例-2
[0160]
将472克a,1528克b(悬浮法san),6克硬脂酸镁,6克ebs,180克c2共混后挤出造粒,组合物性能为:冲击强度278.1j/m,抗张强度32.4mpa,mi1.9g/10min,表面光泽(600)61。
[0161]
比较例-2说明:尽管使用了本发明规定的a、b用量范围,但c2的用量超出本发明的范围后,其对降低产品的光泽仍然有效,但拉伸强度和冲击性能都不友好,也从另一个角度说明c在组合物中用量存在适宜的范围,添加过量时会劣化组合物的相容性,导致冲击强度的下降。
[0162]
比较例-3
[0163]
将1000克a,1000克b(悬浮法san),6克硬脂酸镁,6克ebs,70克c2共混后挤出造粒,组合物性能为:冲击强度341.1j/m,抗张强度29mpa,mi0.3g/10min,表面光泽(600)61。
[0164]
比较例-3说明:使用高于本发明规定的a用量范围,同时使用本发明适宜的c2用量,制品的冲击强度、降低光泽的效果比较理想,但拉伸强度却无法达到本发明需要的效果。
[0165]
比较例-4
[0166]
将500克a,1500克b(悬浮法san),6克硬脂酸镁,6克ebs,共混后挤出造粒,组合物性能为:冲击强度272.3j/m,抗张强度46.2mpa,mi1.3g/10min,表面光泽(600)95。
[0167]
比较例4说明:组合物只有a和b,不使用本发明的光泽调节剂c,无法获得本发明所说的光泽降低的亚光化产品。
[0168]
以上所述实施例仅表达了本发明的几种实施方式,其描述较为具体和详细,但并不能因此而理解为对发明专利范围的限制。应当指出的是,对于本领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干变形和改进,这些都属于本发明的保护范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。