1.本发明涉及阿福拉纳合成领域,尤其是涉及一种阿福拉纳的合成方法。
背景技术:
2.阿福拉纳(afoxolaner),商品名尼可信(nexgard),由勃林格殷格翰公司研制生产,属于异噁唑啉类药物。其作用原理为,通过抑制节肢动物gaba氯离子通道,从而阻断氯离子从突触前膜到突触后膜的传递,导致昆虫神经元活性增加,兴奋过度死亡。该药于2017年8月在中国获批上市,为国内首款兼杀蜱虫和跳蚤两种寄生虫的犬用口服驱虫药,能够满足宠物主人“内外兼顾”的需求,犬类动物每月仅需服用一次,就可以驱除和预防心丝虫、蛔虫、钩虫、鞭虫、跳蚤、蜱虫等6大类体内外常见寄生虫。中国农业部于2018年批准进口药物阿福拉纳米尔贝肟咀嚼片作为用于治疗犬跳蚤、蜱感染,同时用于预防犬心丝虫感染和治疗胃肠道线虫感染。目前,该药物在宠物驱虫药领域中市场发展良好。
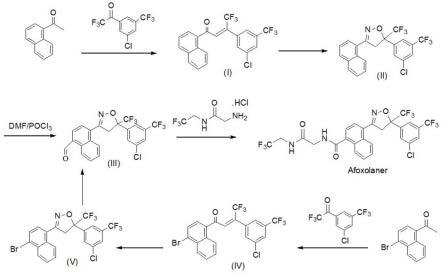
3.现有技术中,阿福拉纳的合成重点在于,异噁唑环的生成及侧链的引入。其中,侧链可以在异噁唑环合成之前引入,也可在异噁唑合成之后引入。目前,已有多篇文献公开了在异噁唑环合成之前引入侧链,具体是选择先在相应的中间体中引入侧链(其技术路线如说明书附图1所示),先制得中间体a或c或d;然后异噁唑环的合成采用两个相应的酮a、b先进行缩合,再与羟胺反应制得阿福拉纳(如pct专利wo2013021949a1);或者通过进行[3 2]成环反应制得阿福拉纳(如中国专利cn109879826a、中国专利cn111675667a、中国专利cn112457267a)。具体合成路线见说明书附图1。
[0004]
但是,发明人经研究发现,在异噁唑环合成之前引入侧链的方法存在有,原料不易获得,价格昂贵,且降低了分子极性的缺陷;同时,在后续环合反应的后处理过程中,成品杂质较多且不易于纯化,杂质容易被带入成品中,而使得成品的精制更加困难,生产难度大,能耗高,成本较高。
[0005]
由此,克服现有技术中前述的阿福拉纳合成工艺中存在的明显不足,研发一条适合工业化放大生产的合成路线,对于阿福拉纳的产业化尤其重要。
技术实现要素:
[0006]
为解决现有技术中存在的技术问题,本发明提供一种阿福拉纳的合成方法,能够克服现有阿福拉纳合成方法中存在的原料价格昂贵、不易获得,环合反应后处理困难、成品杂质较多、难以纯化等技术难题,合成方法中采用的原料廉价易得,后处理纯化简单,生产能耗低,适合规模化工业生产。
[0007]
为解决以上技术问题,本发明采取的技术方案如下:一种阿福拉纳的合成方法,通过中间体iii与2-氨基-n-(2,2,2-三氟乙基)乙酰胺盐酸盐、过氧化氢叔丁醇,在30℃温度条件下,经氧化-缩合制得阿福拉纳;具体的,所述中间体iii和过氧化氢叔丁醇投入至混合液中,30℃温度条件下,搅拌反应15h后,经萃取、减压浓缩、结晶、干燥,制得阿福拉纳;
所述中间体iii、2-氨基-n-(2,2,2-三氟乙基)乙酰胺盐酸盐、过氧化氢叔丁醇的摩尔份比值为1:1:1.1。
[0008]
所述混合液为,将cuso4·
5h2o、2-氨基-n-(2,2,2-三氟乙基)乙酰胺盐酸盐、三乙胺、caco3和乙腈混合均匀制得;所述cuso4·
5h2o、2-氨基-n-(2,2,2-三氟乙基)乙酰胺盐酸盐、三乙胺、caco3的摩尔份比值为0.05:1:2:1.1。
[0009]
所述中间体iii,具有如下的结构式:;所述中间体iii通过第一方法或第二方法制备获得;所述第一方法为,将中间体ii溶解于dmf中,在0℃温度条件下,滴加三氯氧磷,滴加完成后,在0℃温度条件下,保温反应10h后,经洗涤、乙酸乙酯萃取、无水硫酸钠干燥、减压浓缩、结晶、干燥,制得中间体iii;所述中间体ii,具有如下的结构式:;所述第一方法中,中间体ii、三氯氧磷的摩尔份比值为3:4。
[0010]
所述第二方法为,将中间体v溶解于四氢呋喃中,然后投入镁屑、碘,升温至40℃,保温搅拌均匀;然后滴入n,n-二甲基甲酰胺的四氢呋喃溶液,保温反应后,经洗涤、萃取、无水硫酸钠干燥、减压浓缩、结晶、干燥,制得中间体iii;所述中间体v,具有如下的结构式:
;所述第二方法中,中间体v、镁屑的摩尔份比值为1.24:1.5。
[0011]
所述中间体ii的制备方法为,采用1-萘乙酮、3'-氯-5'-三氟甲基-2,2,2三氟苯乙酮,在溶剂甲苯中,在三乙胺的催化下,50℃温度下,保温搅拌反应10h,经洗涤、减压干燥,制得中间产物;将中间产物、氯化亚砜溶解于甲苯中,在60℃温度条件下,滴入吡啶,滴加完成后,保温搅拌反应2h后,经洗涤、减压浓缩,制得中间体i;然后将中间体i溶解于甲苯中,然后加入四丁基溴化铵,在0℃温度条件下,搅拌反应1h后;然后滴入第一溶液,滴加完成后,在40℃温度条件下,保温反应3h后,调节ph值至6-7,然后经乙酸乙酯萃取、减压浓缩,制得中间体ii;所述第一溶液为,氢氧化钠、纯化水、50%羟胺水溶液的混合溶液。
[0012]
所述中间体i,具有如下的结构式:;所述1-萘乙酮、3'-氯-5'-三氟甲基-2,2,2三氟苯乙酮的摩尔份比值为1:1。
[0013]
所述中间体v的制备方法为,采用l-(4-溴-l-萘基)乙酮、1-[3-氯-5-(三氟甲基)苯基]-2,2,2-三氟乙酮,在溶剂四氢呋喃中,在二异丙胺锂催化下,0℃温度下,保温反应1.5h后,经乙酸乙酯萃取、无水硫酸钠干燥、减压浓缩、结晶、干燥,制得中间产物;将中间产物溶解于甲苯中,然后加入氯化亚砜,60℃温度下,保温搅拌反应2h后,经乙酸乙酯萃取、无水硫酸钠干燥、减压浓缩,制得中间体iv;然后中间体iv与硫酸羟胺、碳酸钠、水,在溶剂异丙醇中,40℃温度条件下,保温搅拌反应6h后,经乙酸乙酯萃取、无水硫酸钠干燥、减压浓缩,制得中间体v。
[0014]
所述中间体iv,具有如下的结构式:;所述l-(4-溴-l-萘基)乙酮、1-[3-氯-5-(三氟甲基)苯基]-2,2,2-三氟乙酮的摩尔份比值为1:1。
[0015]
与现有技术相比,本发明的有益效果为:(1)本发明的阿福拉纳的合成方法,提供一种全新的阿福拉纳合成路线,通过创造性的设计关键中间体ii和关键中间体v,并采用关键中间体ii或关键中间体v经甲酰化反应制得关键中间体iii后,关键中间体iii与2-氨基-n-(2,2,2-三氟乙基)乙酰胺盐酸盐经氧化-缩合制得阿福拉纳;有效克服现有的在异噁唑环合成之前引入侧链生产阿福拉纳的工艺中,原料价格昂贵、不易获得、降低分子极性的缺陷;关键中间体ii和关键中间体v均能够通过廉价、易得的原料合成;同时,在后续的环合反应中,杂质含量低,后处理简单,产品易于纯化,纯化所需能耗低,适合大规模化工业生产。
[0016]
(2)本发明的阿福拉纳的合成方法,关键中间体ii收率可达87.5%,关键中间体v收率可达83.2%,关键中间体iii收率可达88.6%,最终制得的阿福拉纳hplc纯度可达99.53%,收率可达83%。
附图说明
[0017]
图1为背景技术中的现有合成路线示意图。
[0018]
图2为本发明实施例1-3的反应原理示意图。
[0019]
图3为本发明实施例3制得的阿福拉纳的hplc检测图谱。
具体实施方式
[0020]
为了对本发明的技术特征、目的和效果有更加清楚的理解,现说明本发明的具体实施方式。
[0021]
本发明通过创造性的设计关键中间体ii和关键中间体v;关键中间体ii和关键中间体v均可分别通过甲酰化反应制得关键中间体iii;最后关键中间体iii与2-氨基-n-(2,2,2-三氟乙基)乙酰胺盐酸盐氧化-缩合制得阿福拉纳。
[0022]
具体的,本发明的阿福拉纳合成方法,选用1-萘乙酮或1-(4-溴-1-萘基)乙酮为起始原料,与3,5-二氯-三氟苯乙酮进行羟醛缩合(aldol缩合),得到中间体i或iv;然后中间体i或iv与羟胺环合反应得到关键中间体ii或关键中间体v;关键中间体ii或关键中间体v
再经甲酰化反应,得到关键中间体iii;最后关键中间体iii与2-氨基-n-(2,2,2-三氟乙基)乙酰胺盐酸盐、氧化剂反应,得到阿福拉纳。具体合成路线见说明书附图2。
[0023]
实施例1实施例1的技术路线为,采用1-萘乙酮为起始原料,与3,5-二氯-三氟苯乙酮进行羟醛缩合(aldol缩合),得到中间体i;然后中间体i与羟胺环合反应得到关键中间体ii;关键中间体ii再经甲酰化反应,得到关键中间体iii。具体方法如下:1、合成中间体i(缩合反应)所述中间体i的结构式如下:;称取1.70g(10.0mmol)1-萘乙酮、2.76g(10.0mmol)3'-氯-5'-三氟甲基-2,2,2三氟苯乙酮、10ml甲苯和0.20g三乙胺加入50ml烧瓶中,然后于50℃温度条件下,搅拌反应10h;滤出反应生成的固体,用5ml甲苯洗涤后,减压干燥得3.6g白色固体。取所得白色固体3.0g、10ml甲苯和1.0g氯化亚砜加入烧瓶中,加热至60℃,保温并缓慢滴加0.6g吡啶,滴加完成后,在60℃温度条件下,搅拌反应2h;然后自然冷却至室温,加入冰水,采用氢氧化钠水溶液(0.5mol/l)洗涤有机相;然后在50℃温度条件下,真空度0.07mpa下,减压浓缩,得3.52g中间体i(收率82.2%),中间体i的核磁共振结果为:1h nmr (dmso-d6)δ9.30(d, 1h), 8.35-8.32(m, 2h),7.97-7.81(m, 5h), 7.53(m, 1h),7.35(d, 1h),7.29(d, 1h)。
[0024]
2、合成中间体ii(环合反应)所述中间体ii的结构式如下:
;将1.71g(4.0mmol)中间体i溶解于10ml甲苯中,并添加90mg(0.3mmol)四丁基溴化铵,在0℃温度条件下,搅拌反应1h,制得反应混合物。然后将第一溶液逐滴滴加到反应混合物中,在40℃温度条件下,保温反应3h。
[0025]
其中,第一溶液为,采用0.193 g(2.2mmol)氢氧化钠、1ml纯化水和0.528g(8.0mmol)50%羟胺水溶液混合制得的溶液。
[0026]
经tlc检测反应完全,冷却后,采用1mol/l的盐酸调节ph值至6-7,然后用20ml乙酸乙酯萃取3次,合并有机层,然后在50℃温度条件下,真空度0.07mpa下,减压浓缩,制得1.55g中间体ii,收率87.5%。中间体ii的核磁共振结果为:1h nmr (dmso-d6)δ8.38-8.30(m, 2h), 7.92-7.36(m, 8h),3.27(d, 1h),3.02(d, 1h)。
[0027]
3、合成中间体iii(甲酰化反应)所述中间体iii的结构式如下:; 称取1.33g(3mmol)中间体ii溶解于10mldmf中,在0℃温度条件下,缓慢加入0.61g(4mmol)三氯氧磷,加入完成后,0℃温度条件下,反应10h,反应结束后,将反应液投入至50ml冰水中,再用300ml水和100ml饱和nahco3进一步稀释,搅拌5h,然后采用50ml乙酸乙酯提3次,合并有机相,加入2g无水硫酸钠干燥3小时,抽滤,滤液在50℃温度条件下,真空度0.07mpa下,减压浓缩至总体积1/3,得浓缩物。搅拌条件下,将50ml正己烷滴加至浓缩物中,滴加完成后,继续搅拌,结晶析出,过滤,干燥,制得1.25g白色固体,即中间体iii,收率88.4%。中间体iii的核磁共振结果为:1h nmr (dmso-d6)δ9.92 (s, 1h),9.30(d, 1h),8.47-8.42(m, 2h),7.96-7.38(m, 6h), 3.27(d, 1h), 3.02(d, 1h)。
[0028]
实施例2实施例2的技术路线为,采用1-(4-溴-1-萘基)乙酮为起始原料,与3,5-二氯-三氟苯乙酮进行羟醛缩合(aldol缩合),得到中间体iv;然后中间体iv与羟胺环合反应得到关键中间体v;关键中间体v再经甲酰化反应,得到关键中间体iii。具体方法如下:1、合成中间体iv(缩合反应)所述中间体iv的结构式如下:;取5.1ml二异丙胺锂的四氢呋喃溶液(二异丙胺锂浓度为2mol/l,10.2mmol),在0℃温度条件下,加入至8ml四氢呋喃中,制得一次混合物。将l-(4-溴-l-萘基)乙酮2.49g(10mmol)溶解于4ml四氢呋喃中,然后逐滴滴加到一次混合物中,滴加完成后,于0℃温度下,搅拌30分钟,得二次混合物。然后将1-[3-氯-5-(三氟甲基)苯基]-2,2,2-三氟乙酮2.76g(10mmol)溶于4ml四氢呋喃中,逐滴滴加到二次混合物中,滴加过程中,控制滴加速率使反应混合物的温度不超过-5℃。滴加完成后,继续搅拌1h。然后将反应混合物投入至20ml盐酸中,用30ml乙酸乙酯萃取2次,合并有机相,用无水硫酸钠干燥,抽滤,滤液在50
±
5℃温度条件下,真空度0.07mpa下,减压浓缩,得浓缩物;将浓缩物中加入至20ml乙酸乙酯/正己烷(v:v=1:9)中,搅拌结晶,过滤、干燥得4.35g白色固体,收率85.9%,熔点:74-75℃,即1-(4-溴代萘-1-基)-3-(3-氯-5-(三氟甲基)苯基)-4,4,4-三氟-3-羟基丁烷-1-酮。
[0029]
将2.53g(5mmol)前述产物溶解于10ml甲苯,然后加入1.2g(10mmol)氯化亚砜,在60℃温度条件下,搅拌反应2h,冷却至室温后,加入10ml稀盐酸(1mol/l),然后用25ml乙酸乙酯萃取2次,合并有机层,加入2g无水硫酸钠干燥2小时,抽滤,滤液在50℃温度条件下,真空度0.07mpa下,减压浓缩,得2.23g油状物,收率85.6%,即1-(4-溴代萘-1-基)-3-(3-氯-5-(三氟甲基)苯基)-4,4,4-三氟-2-丁烯-1-酮(中间体iv)。中间体iv的核磁共振结果为:1h nmr (dmso-d6)δ9.29(d, 1h),8.28-8.23(m, 2h),8.07(d, 1h),7.92(m, 1h),7.86(s, 1h),7.65-7.53(m, 2h),7.36(d, ih),7.29(d, 1h)。
[0030]
2、合成中间体v(环合反应)所述中间体v的结构式如下:
;将1.53g中间体iv(3mmol)溶解于10ml异丙醇中,然后加入0.74g硫酸羟胺(4.5mmol)、0.95g碳酸钠(9mmol)、5ml水,于40℃温度条件下,搅拌反应6h。经tlc监测反应完毕,加入20ml水,用25ml乙酸乙酯萃取2次,合并有机层,加入2g无水硫酸钠干燥2小时,抽滤,滤液在50℃温度条件下,真空度0.07mpa下,减压浓缩,得1.31g 4-{5-[3-氯-5-(三氟甲基)苯基]-5-三氟甲基-4,5-二氢异噁唑-3-基}萘-1-甲醛(中间体v),收率83.2%。中间体v的核磁共振结果为:1h nmr (dmso-d6)δ8.26-8.15(m, 3h),7.90(d, 1h),7.75(m, 1h),7.62(d, 1h),7.59(m, 1h),7.46(d, 1h),7.38(d, 1h),3.27(d, 1h),3.02(d, 1h)。
[0031]
3、合成中间体iii(甲酰化反应)所述中间体iii的结构式如下:;取0.65g中间体v(1.24mmol),溶于10ml四氢呋喃中,然后加入0.036g镁屑(1.5mmol)、5mg碘,搅拌条件下,升温至40℃后,制得反应液。将n,n-二甲基甲酰胺0.1g溶于2ml四氢呋喃中,滴加至反应液中,经tlc监测反应完毕后,滴加2ml稀盐酸(1mol/l),继续搅拌1h,然后采用20ml二氯甲烷萃取2次,合并有机层,加入1g无水硫酸钠干燥,抽滤,滤液于35℃温度条件下,真空度0.07mpa下,减压浓缩至总体积1/3,得浓缩物。搅拌条件下,将50ml正己烷滴加至浓缩物中,滴加完成后,继续搅拌,结晶析出,过滤、干燥,得到0.52g类白色固体,即4-{5-[3-氯-5-(三氟甲基)苯基]-5-三氟甲基-4,5-二氢异噁唑-3-基}萘-1-甲醛(中间体iii),收率88.6%。
[0032]
实施例3合成阿福拉纳(氧化-缩合)采用实施例1或实施例2制得的中间体iii,与2-氨基-n-(2,2,2-三氟乙基)乙酰胺
盐酸盐、氧化剂反应,得到阿福拉纳。具体方法如下:将cuso4·
5h2o(12.5mg,0.05mmol)、2-氨基-n-(2,2,2-三氟乙基)乙酰胺盐酸盐(193mg,1.0mmol)、三乙胺(202mg,2.0mmol)、caco3(110mg,1.1mmol)和乙腈(5ml)投入到单口烧瓶中,氮气气氛保护下,加入中间体iii(471mg,1mmol)和过氧化氢叔丁醇(70%水溶液,0.16ml,1.1mmol),在30℃温度条件下,搅拌反应15h后,30ml乙酸乙酯萃取2次,合并有机相,于35℃温度条件下,真空度0.07mpa下,减压浓缩,浓缩液在乙酸乙酯/甲基叔丁基醚(体积比为8:2)体系中结晶,制得522mg阿福拉纳,hplc纯度99.53%,收率83%。核磁共振结果为:1h nmr(400mhz,cdcl3)δ9.35(m, 2h),8.39(m, 2h), 7.57-7.46(m, 5h),7.12(m, 1h),6.84(m, 1h),4.24(d, 2h),4.10(d, 1h),4.00(m, 2h),3.76(m, 1h)。
[0033]
除非另有说明,本发明中所采用的百分数均为质量百分数。
[0034]
最后应说明的是:以上所述仅为本发明的优选实施例而已,并不用于限制本发明,尽管参照前述实施例对本发明进行了详细的说明,对于本领域的技术人员来说,其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分技术特征进行等同替换。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。