1.本发明属于基因组学和分子生物学技术领域,特别涉及癌诊筛查模型及其构建方法和装置,尤其涉及一种肝细胞癌筛查模型及其构建方法和装置。
背景技术:
2.原发性肝细胞癌(hepatocellular carcinoma,hcc)是一类常见的恶性肿瘤,在我国的发病率和死亡率位居全球前列。目前,中国大多数肝细胞癌患者初诊时已经是中晚期甚至终末期,无法实施根治性手术,严重影响肝细胞癌预后,提高早期肝细胞癌检出率并实施个性化治疗,可明显降低肝细胞癌死亡率。然而早期肝细胞癌患者症状并不明显,肝脏超声、血清alpha-fetoprotein(afp)检查及pivka-ii检查作为常用的肝细胞癌的筛查诊断技术,对早期肝细胞癌的敏感性比较低。因此,迫切需要开发一种敏感、可靠、微创的检测方法,发现早期肝细胞癌,以便及时干预。
3.血浆细胞游离dna(plasma cell free dna,cfdna)是指细胞坏死或凋亡后释放到血浆中的降解dna片段。在癌症患者中,cfdna的一部分来源于肿瘤细胞,也被称为循环肿瘤dna(ctdna)。下一代测序(next-generation sequencing,ngs)可以分析ctdna携带的多种肿瘤特异性信号,尤其是dna甲基化修饰,这一具有反映肿瘤早期变化特性的优势生物标志物。
4.近年来,使用ctdna相关的生物标志物进行临床转化研究,并结合抽血等形式的液体活检的无创手法用于肿瘤的早期诊断是肿瘤早筛研究的热点,在获得ctdna携带的肿瘤特异性信号后,通常结合机器学的方法,利用该信号作为特征建立的癌症早筛模型。融合模型属于机器学习模型中的集成学习模型,该算法通过构建并结合多个学习器来完成分类任务,这种方法常可获得比单一学习器显著优越的泛化性能,得到一个表现更好,更具有鲁棒性的分类器,有助于解决关键的计算问题,已运用到人脸识别、语言识别等领域。然而,将该技术应用在早期肝细胞癌筛查方面还未见报道。
技术实现要素:
5.本发明的目的是提供一种肝细胞癌筛查模型的构建方法和装置,可以高效区分肝细胞癌样品和非肝细胞癌样品,本方法敏感性高、特异性高、无创伤性。
6.为此,本发明技术方案如下:
7.第一方面,本发明提供一种肝细胞癌的筛查模型的构建方法,所述方法利用基于机器学习的融合模型。
8.优选地,所述构建方法至少包括以下步骤:
9.步骤一:采集已确诊为肝细胞癌的患者数据和非肝细胞癌者数据的dna甲基化数据;
10.步骤二:对已确诊为肝细胞癌的患者数据和非肝细胞癌的数据的dna甲基化数据进行分析,获得符合要求的每条测序序列位点对应的甲基化信息数据;
11.步骤三:利用上一步骤的甲基化信息数据,构建、优化和测试融合模型。
12.优选地,步骤一所述非肝细胞癌者包括正常人群、肝硬化人群和肝炎人群的dna甲基化数据;
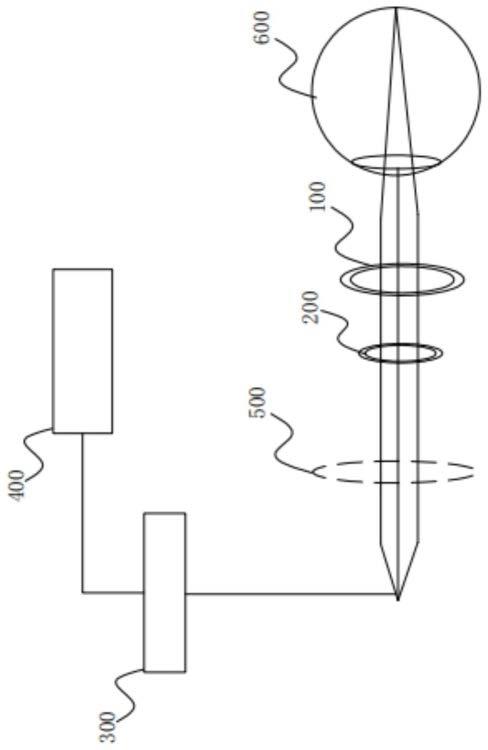
13.优选地,步骤一至少包括以下步骤:
14.101)抽取受检者外周血、离心、分离血浆、提取血浆游离dna;所述受检者为已确诊为肝细胞癌的患者和非肝细胞癌者;
15.102)对血浆游离dna进行dna甲基化测序,构建甲基化测序文库;
16.103)将甲基化测序文库与选定的肝细胞癌特征标志物组合探针杂交富集扩增,得到最终文库进行上机测序,分析受检者的dna甲基化数据。
17.现有的用于构建甲基化测序文库的dna甲基化测序的实验方法包括但不限于:重亚硫酸盐测序法,酶学甲基化转化测序(em-seq)法,taps法等,上述方法在本发明中均可使用。
18.优选地,步骤二至少包括以下步骤:
19.201)dna甲基化数据下机后,使用数据处理软件将步骤一所述的dna甲基化数据进行质控、比对、排序和去重;
20.202)设定适当条件,过滤不符合要求的测序序列(过滤条件包括但不限于:测序质量,序列长度等),获得符合要求的每条测序序列位点对应的甲基化信息数据。
21.具体的,使用fastp(0.21.0)对原始fastq文件进行处理,使用默认参数去掉illumina特定接头,去掉低质量序列的参数为-u 20和-q 20。将处理之后的clean fastq数据利用bismark(0.23.0)进行甲基化比对。使用deduplicate_bismark进行去重处理。bismark生成的比对文件中包括了每条reads的每个碱基的甲基化信息,统计每条read中chg和chh的数量,去除chg和chh数量和大于等于3的reads。最后利用bismark_methylation_extractor(bismark)得到甲基化注释信息。
22.优选地,步骤三至少包括以下步骤:
23.301)将步骤二测得的甲基化信息数据进行分类,区分为已确诊为肝细胞癌的患者数据和非肝细胞癌的数据,分别加入标签列,标注为肝细胞癌和非肝细胞癌;
24.302)将分类好的数据按照70%和30%的比例,分别划分为训练集和测试集;
25.303)构建融合模型,输入训练集的特征数据和标签对融合模型进行训练和优化,得到最终模型;
26.304)将测试集输入最终模型中,使用分类结果对最终模型进行效果评价。
27.优选地,步骤303)所述融合模型分为两层,第一层为两个模型,第二层为一个多项式逻辑回归模型;
28.其中,第一层包括一个肝细胞癌与正常模型,使用训练集中的正常人数据和肝细胞癌数据训练得出,(hcc vs normal,下称“hn模型”)和一个肝细胞癌与肝病模型,使用训练集中的肝病数据和肝细胞癌数据训练得出(hcc vs liver-disease,下称“hl模型”);
29.优选地,第一层的两个模型均采用gdbt(gradient boosting decision tree),即梯度提升迭代决策树算法进行训练;
30.优选地,第二层多项式逻辑回归模型的构建方式为:将第一层的两个模型采用stack方式使用逻辑回归进行模型融合,将融合模型的结果作为对样本的肝细胞癌和非肝
细胞癌的预测;其中,构建第二层模型的过程实现可使用r语言和/或python语言,优选python(v.3.6.7)语言。
31.模型的第一层的算法选用gdbt。其中,所述gdbt算法的具体过程为:
32.1-1)首先,初始化弱分类器:
[0033][0034]
在公式(1)中,n为训练集样本总数量,yi为训练集样本的标签,x为训练集的特征数据;
[0035]
1-2)利用公式(2)计算m棵树的残差r
m,i
,依次进行迭代,其中m=1,2,
…
,m,m为最大迭代次数;i=1,2,
…
,n
[0036][0037]
1-3)对于第m棵回归树的叶子结点区域r
mj
,其中j=1,2,...,jm;jm为第m棵回归树叶子结点的个数,对于jm个叶子结点区域,利用公式(3)计算出最佳残差拟合值c
mj
:
[0038][0039]
1-4)更新分类器,直到获得最终强的分类器fm(x),过程中的计算公式如公式(4)和公式(5)所示:
[0040][0041][0042]
模型第一层的具体构建方法可描述为:分别使用训练集中的正常人和肝细胞癌病人数据、肝病(肝硬化 肝炎)和肝细胞癌病人的数据作为训练数据,按照上述步骤1-1)至1-4)训练得出两个模型,分别称为hn(hcc normal)模型和hl(hcc liver_disease)模型,模型特征工程的方法设定为根据模型对特征权重的打分,选择权重大于0的特征。模型参数使用网格搜索结合交叉验证的方式来进行优化,优化后第一层训练完毕。
[0043]
模型的第二层选用逻辑回归算法构建的模型进行训练,具体的,所述逻辑回归算法可描述为:
[0044]
2-1)首先,构造一个预测函数:
[0045][0046]
2-2)进一步的,为了避免过拟合,将逻辑回归算法中加入l2正则化,具体的过程可描述为:对损失函数j求偏导,迭代直到损失值达到最小,模型收敛。
[0047]
具体如公式(7)和公式(8)所示:
[0048][0049][0050]
模型第二层的具体构建方法可描述为:将模型第一层的两个模型的输出作为特征,以矩阵的形式输入,使用逻辑回归算法的按照上述步骤2-1)至2-2)训练后得到最终的模型。对于第二层模型的参数,也使用网格搜索结合交叉验证的方式来进行优化,优化后第二层训练完毕,形成最终模型。
[0051]
具体的,所述效果评价为:
[0052]
使用步骤三划分得到的测试集输入优化好的最终模型,使用分类结果对最终模型进行效果评价。根据分类结果画出最终模型使用的特征分类效果图;计算出最终模型的敏感性、特异性和准确率;最终模型的roc曲线图和auc的值;以及最终模型和现有检测手法afp和pivka-ii检测结果的比较值。
[0053]
第二方面,本发明提供一种肝细胞癌筛查模型的构建装置,所述构建装置包括:
[0054]
模块一:用于采集已确诊为肝细胞癌的患者数据和非肝细胞癌者数据的dna甲基化数据;
[0055]
模块二:用于对已确诊为肝细胞癌的患者数据和非肝细胞癌的数据的dna甲基化数据进行分析,获得符合要求的每条测序序列位点对应的甲基化信息数据;
[0056]
模块三:用于利用模块二的甲基化信息数据,构建、优化和测试融合模型。
[0057]
第三方面,本发明提供一种肝细胞癌筛查模型,所述肿瘤筛查模型通过本发明第一方面所述的方法构建而成。
[0058]
第四方面,本发明提供一种肝细胞癌筛查装置,所述肝细胞癌筛查装置包括本发明第一方面所述的方法构建的肝细胞癌筛查模型。
[0059]
与现有技术相比,本发明提供的肝细胞癌筛查模型及其构建方法和装置至少具有以下优点:
[0060]
本发明利用下一代测序(generation sequencing,ngs)获得标记物dna甲基化数据,构建机器学习融合模型预测肝细胞癌。构建的模型具有良好的稳定性和鲁棒性,可以高效区分肝细胞癌样品和非肝细胞癌样品,提供了一种敏感性高、特异性高、无创的肝细胞癌检测思路。
附图说明
[0061]
图1为本发明模型构建方法的总技术路线图;
[0062]
图2为实施例1融合模型构建技术路线图;
[0063]
图3为实施例1特征分类效果热图;
[0064]
图4为实施例1融合模型评价roc曲线图;
[0065]
图5为实施例1融合模型和现有检测手法结果比较图。
具体实施方式
[0066]
下面详细描述本发明的实施例,所述实施例的示例在附图中示出,其中自始至终相同或类似的标号表示相同或类似的元件或具有相同或类似功能的元件。下面通过参考附图描述的实施例是示例性的,旨在用于解释本发明,而不能理解为对本发明的限制。
[0067]
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不必须针对的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任何的一个或多个实施例或示例中以合适的方式结合。此外,本领域的技术人员可以将本说明书中描述的不同实施例或示例进行接合和组合。
[0068]
实施例1
[0069]
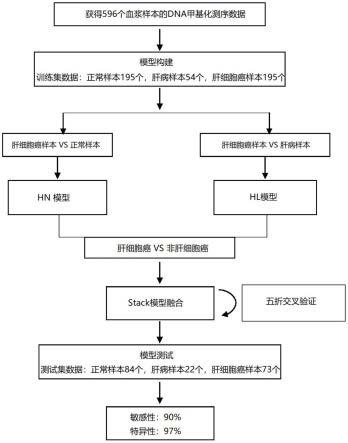
本实施例的融合模型构建技术路线图如图2所示。
[0070]
步骤一:采集dna甲基化数据
[0071]
本实施例共募集596位志愿者,其中肝细胞癌268人,肝病76人,正常人274人,所有样本均采用以下方法处理:
[0072]
将志愿者的外周血分别采集于cfdna保存管(厦门志煊),离心分离血浆,从血浆中提取cfdna。采用vahts universal dna library prep kit for illumina v3(诺唯赞)将cfdna连接带有甲基化修饰的接头,并对连接产物进行酶学转化处理(enzymatic methyl-seq conversion module,neb)。转化产物进行扩增纯化后,用目标探针捕获。经pcr扩增获得捕获文库,最后进行上机测序。
[0073]
步骤二:数据处理方法
[0074]
步骤一的dna甲基化序列测序数据下机后,使用fastp(0.21.0)对原始fastq文件进行处理,使用默认参数去掉illumina特定接头,去掉低质量序列的参数为-u 20和-q 20。将处理之后的clean fastq数据利用bismark(0.23.0)进行甲基化比对。使用deduplicate_bismark进行去重处理。bismark生成的比对文件中包括了每条reads的每个碱基的甲基化信息,统计每条read中chg和chh的数量,去除chg和chh数量和大于等于3的reads。最后利用bismark_methylation_extractor(bismark)得到甲基化注释信息。参数设置为:comprehensive
‑‑
bedgraph
‑‑
counts
‑‑
cytosine_report
‑‑
cx
‑‑
buffer_size 20g
‑‑
parallel 16。
[0075]
步骤三:融合模型构建与优化
[0076]
将步骤二得到的596个志愿者的数据按照临床信息分为两组,一组是包括正常人和肝病患者的非肝细胞癌组,另一组是肝细胞癌组。随后将两组数据随机划分,数据的70%分为训练集(n=417,正常人=195,肝病=54,肝细胞癌=168),用于训练模型,30%划分为测试集(n=179,正常人=84,肝病=23,肝细胞癌=72),用于测试模型,过程是单盲测试。模型构建和优化部分优选使用python(v.3.6.7)语言实现,具体构建过程,首先从scikit-learn(v1.1.0)中调用gradientboostingclassifier的包,分別输入正常人数据、肝细胞癌数据,以及肝病数据和肝细胞癌数据,实现使用gdbt算法训练得到两个模型,即hn模型和hl模型,模型的第一层训练完毕。然后再从scikit-learn(v1.1.0)中调用logisticregression包,将hn模型和hl模型的输出作为特征构建模型的第二层,过程中采
用l2正则化防止模型过拟合,实现使用逻辑回归算法以stack的方式,训练得到最终的模型。对于模型的参数,使用网格搜索结合交叉验证的方式来进行优化,可调整的参数包括:solver,max_iter,c值等。
[0077]
步骤四:模型结果测试
[0078]
1)将步骤三划分得到的测试集数据输入模型,得到测试集每个样本的模型预测结果,接着进行模型效果的计算和画图展示。这部分包括:
[0079]
根据模型使用的肝细胞癌相关甲基化位点特征的值,画出在训练集和测试集中的在非肝细胞癌组和肝细胞癌病人组的特征数据热图,见图3。根据所选特征,可以明显的看出肝细胞癌与非肝细胞癌人群的差异。因此,所述肝细胞癌相关甲基化位点组合,在肝细胞癌病人和正常对照的血浆游离dna中这些位点的甲基化信号具有明显的差别,可以作为检测肝细胞癌的液体活检标记物使用。
[0080]
2)将测试集的实际结果进行比较,计算具体的敏感性和特异性,画出模型的测试集和训练集的roc曲线图,计算auc和置信区间,这部分通过r(v.3.6.0)语言来实施,由proc(v.1.18.0)包计算得出;测试集中将临床信息包括有afp和pivka-ii检测结果的肝细胞癌患者的模型预测结果整理出来,将这两种临床使用的蛋白质检测和模型的检测结果进行准确性的比较。
[0081]
本实施例构建的模型结果计算如下:
[0082]
模型在训练集的五折交叉验证效果为敏感性92%,特异性为94%(auc=0.980,95%ci 0.968-0.991)。在测试集中,敏感性为90%,特异性为97%(auc=0.957,95%ci 0.925-0.989),具体结果见图4。模型在测试集肝细胞癌组中的敏感性随着癌症分期的推进而提高,i期患者的敏感性达到85%(35/41),ii期患者达到89%(8/9),iii-iv期患者达到100%(23/23)。模型在测试集非肝细胞癌组中表现为高特异性,其中肝病患者的特异性为91%,在正常人群中的特异性为99%,具体结果见表1。
[0083]
在测试集的肝细胞癌患者中,模型的预测结果与现有技术常用的血清afp检测(阈值为20ng/ml)进行比较。可以看出,无论是早期(n=50)还是晚期(n=22)肝细胞癌患者,模型的测精度均高于afp,另对于pivka-ii检测结果(阈值为40ng/ml),同样的,对于早期(n=15)和晚期(n=11)肝细胞癌患者,模型具有更高的检测精度,具体结果见图5。
[0084]
表1实施例1融合模型测试集分类效果表
[0085][0086]
综上所述,本发明利用下一代测序(generation sequencing,ngs)获得标记物dna甲基化数据,构建机器学习融合模型预测肝细胞癌。构建的模型具有良好的稳定性和鲁棒性。模型可以高效区分肝细胞癌样品和非肝细胞癌样品,提供了一种敏感性高、特异性高、无创的肝细胞癌检测方法。
[0087]
应该注意到并理解,在不脱离后附的权利要求所要求的本发明的精神和范围的情况下,能够对上述详细描述的本发明做出各种修改和改进。因此,要求保护的技术方案的范围不受所给出的任何特定示范教导的限制。
[0088]
申请人声明,以上内容是结合具体的优选实施方式对本发明所作的进一步详细说明,不能认定本发明的具体实施只局限于这些说明。对于本发明所属技术领域的普通技术人员来说,在不脱离本发明构思的前提下,还可以做出若干简单推演或替换,都应当视为属于本发明的保护范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。