1.本发明涉及含氟中间体的制备,特别涉及3-氟代吡唑羧酸酯的制备方法,以及以3-氟代吡唑羧酸酯为原料制备3-氟代烷基-1-取代吡唑-4-羧酸的方法。
背景技术:
2.含氟吡唑衍生物是常见的医药和农药的中间体,其中,3-二氟甲基-1-甲基吡唑-4-羧酸是一个重要的农药中间体,其下游已商品化的农药就有五种,包括:先正达公司的杀菌剂吡唑萘菌胺、氟唑环菌胺、苯并烯氟菌唑,巴斯夫公司的杀菌剂氟唑菌酰胺,以及拜尔公司的杀菌剂联苯吡菌胺。目前,这几种农药市场需求持续增长,3-二氟甲基-1-甲基吡唑-4-羧酸作为上述农药的关键中间体,其合成工艺也是当下的研究热点,现有的制备方法依据原料归结为以下几类:
3.一、二氟乙酰乙酸乙酯路线
4.先正达公司专利ep1997808a公开了以二氟乙酰乙酸乙酯为原料,与原甲酸三乙酯、醋酸酐反应生成中间体,之后与甲基肼环合,最后经水解获得3-二氟甲基-1-甲基吡唑-4-羧酸的方法,该方法步骤简单,三废量少,但是原料昂贵、不易得,且环合产物的异构体含量高达11%。
5.二、酰卤路线
6.拜耳公司专利us2006252944a公开了以酰卤和二烷基氨基丙烯酸酯在碱存在下反应得到中间体,之后和甲基肼环合,最后经水解得到3-二氟甲基吡唑甲酸的方法,此方法虽然路线短,但其酰化步骤需要使用碱作为缚酸剂,且环合步骤的异构体含量仍然有12.7%,此外该制备工艺对设备要求高。
7.三、其他路线
8.索尔维公司专利wo2017129759a公开了以乙烯基乙醚和三氯乙酰氯合成中间体,之后经胺化、二氟乙酰化、与甲基肼的苯甲醛腙环化、水解等步骤合成二氟甲基吡唑甲酸的方法,但该方法步骤长、收率低,三废量大,不适宜产业化生产。
9.先正达公司专利ep2008996a公开了以二氟丙酮、甲基肼、三氯氧磷为原料三步合成二氟甲基吡唑甲酸的方法,但该方法收率低,仅53%,同时会产生大量含磷和dmf的废水,且原料二氟丙酮价格昂贵,不适于产业化生产。
10.巴斯夫公司专利us2010184994a公开了以1-氨基-1,1,2,2-四氟乙烷与乙氧基丙烯酸乙酯为原料,在路易斯酸三氟化硼乙醚的催化下合成亚胺盐离子,再与甲基肼环化,最后经碱水解合成二氟甲基吡唑甲酸的方法,但该方法收率仅为25%,不适合工业化生产。
11.综上所述,目前含氟取代吡唑甲酸的制备工艺,或环合中间体异构体含量高,最低在10.9~12.7%左右;或产品收率低,基本在25~65%左右;或原料不易得、成本高;或工艺复杂、三废量大等,因此,需要开发一种新的含氟取代吡唑甲酸的制备工艺。
技术实现要素:
12.为了解决上述技术问题,本发明提出了一种产物选择性高、收率高、反应条件温和,适于产业化生产的3-氟代吡唑羧酸酯的制备方法以及3-氟代烷基-1-取代吡唑-4-羧酸的制备方法。
13.本发明的目的是通过以下技术方案实现的:
14.一种式(5)所示3-氟代吡唑羧酸酯的制备方法,包括以下步骤:
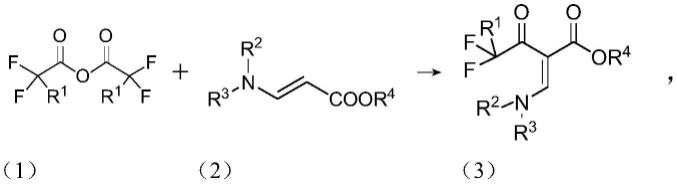
15.a1.酰化步骤:下式(1)所示氟代乙酸酐和式(2)所示3-氨基丙烯酸酯在非质子有机溶剂中反应获得式(3)所示中间体,反应式如下:
[0016][0017]
式中,r1选自h或f;r2、r3、r4独立地选自c
1-c6烷基、c
1-c6卤代烷基、c
2-c6链烯基、c
3-c
10
环烷基、苄基、被取代基取代的c
3-c
10
环烷基或苄基,所述取代基选自卤素、氰基、硝基、c
1-c4烷基、c
1-c4卤代烷基、烷氧基或卤代烷氧基,或r2、r3与其连接的氮原子形成5-10元杂环基,所述5-10元杂环基还可包括o、n、s;
[0018]
a2.环化步骤:所述酰化步骤的反应液直接与下式(4)所示肼类化合物环合生成下式(5)所示3-氟代吡唑羧酸酯,反应式如下:
[0019][0020]
式中,r5选自c
1-c6烷基、c
1-c6卤代烷基、c
2-c6链烯基、c
3-c
10
环烷基、苄基、被取代基取代的c
3-c
10
环烷基或苄基,所述取代基选自卤素、氰基、硝基、c
1-c4烷基、c
1-c4卤代烷基、烷氧基或卤代烷氧基。
[0021]
作为优选,r2、r3、r4、r5独立地选自甲基或乙基。
[0022]
更为优选地,r5选自甲基。
[0023]
本发明所述酰化步骤中,所述氟代乙酸酐选自二氟乙酸酐或三氟乙酸酐。所述非质子有机溶剂不与水互溶,选自苯、甲苯、二甲苯、氯苯、二氯苯、二氯甲烷、二氯乙烷、氯仿、四氯化碳、正己烷、环己烷、石油醚中的至少一种,优选甲苯或二氯甲烷。
[0024]
本发明所述酰化步骤,既可以在无缚酸剂下进行,也可以在缚酸剂作用下进行,所述缚酸剂选自三乙胺、三甲胺、二异丙基乙基胺、三正丙基胺、三正丁基胺、三正己基胺、三环己基胺、n-甲基环己基胺、n-甲基吡咯烷、n-甲基哌啶、n-乙基哌啶、n,n-二甲基苯胺、n-甲基吗啉、吡啶、2-甲基吡啶、3-甲基吡啶、4-甲基吡啶、2-甲基-5-乙基吡啶、2,6-二甲基吡啶、2,4,6-三甲基吡啶、4-二甲基氨基吡啶中的至少一种,优选三乙胺或吡啶。缚酸剂的加入对酰化步骤的反应速率提升具有积极作用。
[0025]
但本发明人经研究发现,当酰化步骤中加入缚酸剂时,反应产生的二氟醋酸盐/三氟醋酸盐会增加环化步骤中5-氟代吡唑羧酸酯的异构体,故,需要采用水洗等方法除去反应产生的醋酸盐。也即,酰化步骤中加入缚酸剂后,酰化步骤的反应液经水洗后参与环化步骤。
[0026]
而当不采用缚酸剂时,反应产生的为二氟醋酸/三氟醋酸,其有利于降低环化步骤中异构体的含量,可提高产品选择性。因此,当不采用缚酸剂时,无需对酰化步骤的反应液进行处理,直接进行环化步骤即可。
[0027]
本发明所述酰化步骤中,所述式(1)所示氟代乙酸酐和式(2)所示3-氨基丙烯酸酯的摩尔比为0.8~1.5:1,反应温度为-30~30℃。作为优选,氟代乙酸酐和3-氨基丙烯酸酯的摩尔比为0.9~1.1:1,反应温度为-10~10℃。当采用缚酸剂时,所述缚酸剂与式(2)所示3-氨基丙烯酸酯的摩尔比为1~1.5:1。
[0028]
本发明所述环化步骤的反应液中包括3-氟代吡唑羧酸酯及其同分异构体5-氟代吡唑羧酸酯,可采用重结晶获得3-氟代吡唑羧酸酯,具体操作如下:
[0029]
酰化步骤的反应液滴入肼类化合物和非质子有机溶剂的混合液中,环化反应结束后蒸除非质子有机溶剂,获得氟代吡唑羧酸酯粗品;
[0030]
采用非极性溶剂对氟代吡唑羧酸酯粗品进行回流重结晶,除去异构体,获得纯化后的3-氟代吡唑羧酸酯,纯度大于99%。所述非极性溶剂选自石油醚、正己烷、环己烷、正庚烷、甲基环己烷中的至少一种,优选正己烷或正庚烷。
[0031]
进一步地,所述非极性溶剂的用量与氟代吡唑羧酸酯粗品质量比为0.5~5:1,该条件下,获得的产品纯度和收率最优。
[0032]
本发明所述环化步骤中,式(4)所示肼类化合物的摩尔用量为式(3)所示中间体理论量的0.9~2倍,反应温度为-50~20℃。作为优选,肼类化合物的摩尔用量为式(3)所示中间体理论量的用量0.95~1.2倍,反应温度-20~0℃。
[0033]
本发明还提供一种式(i)所示3-氟代烷基-1-取代吡唑-4-羧酸的制备方法,所述制备方法包括:
[0034]
b1.水解步骤:上述任一制备获得的3-氟代吡唑羧酸酯在无机碱溶液作用下发生水解,并经酸化获得下式(i)所示3-氟代烷基-1-取代吡唑-4-羧酸:
[0035][0036]
所述无机碱溶液中的无机碱选自选自氢氧化钠、氢氧化钾、氢氧化锂、氢氧化钙、碳酸钠或碳酸钾中的至少一种,无机碱的质量含量为5~50%,优选无机碱为氢氧化钠或氢氧化钾,质量含量为10~30%。
[0037]
本发明所述水解步骤在水相或水与非水溶剂的混合体系中进行,所述非水溶剂选自苯、甲苯、二甲苯、氯苯、二氯苯、二氯甲烷、二氯乙烷、氯仿、四氯化碳、正己烷、环己烷、石油醚、甲醇、乙醇或丙醇中的至少一种,优选乙醇或甲苯。
[0038]
本发明所述水解步骤中,所述3-氟代吡唑羧酸酯与无机碱的摩尔比为1:1.0~2.0,反应温度为10~100℃,优选摩尔比为1:1.0~1.3,反应温度为30~60℃。
[0039]
与现有技术相比,本发明具有的有益效果为:
[0040]
1.本发明采用氟代乙酸酐作为氟化试剂,无需缚酸剂即可高收率、高选择性获得产品,环化反应异构体含量大大降低,工艺简单、后处理方便,适宜工业化生产。
[0041]
2.本发明水解步骤可无溶剂反应,不仅减少溶剂用量,经济环保,还利于提高取代吡唑羧酸的收率。
[0042]
本发明3-氟代吡唑羧酸酯可通过一锅法制备,酰化步骤反应液无需处理即可用于环化步骤,操作简单、三废少。
具体实施方式
[0043]
下面结合具体实施例来对本发明进行进一步说明,但并不将本发明局限于这些具体实施方式。本领域技术人员应该认识到,本发明涵盖了权利要求书范围内所可能包括的所有备选方案、改进方案和等效方案。
[0044]
实施例1
[0045]
本实施例提供3-(二氟甲基)-1-甲基-1h-吡唑-4-羧酸的制备方法,具体步骤如下:
[0046][0047]
s1.酰化步骤:将71.5g(0.5mol)3-(n,n-二甲胺基)丙烯酸乙酯加入到1l三口瓶中,加入甲苯214.5g,开启搅拌,冷却降温至-5℃,向三口瓶中滴加91.4g(0.525mol)二氟乙酸酐,滴加过程中控制温度在-5~0℃之间,滴加完毕后于0℃下保温反应1h,获得酰化反应液;
[0048]
s2.环化步骤:将60.4g(0.525mol)40%的甲基肼水溶液和71g甲苯混合加入1l三口瓶中,搅拌冷却降温到-10℃,缓慢滴加酰化反应液,控制温度-10℃,滴完保温搅拌1小时,之后升温到室温反应1小时。
[0049]
反应液静置分液,有机层水洗一次后减压蒸除甲苯,得到3-二氟甲基吡唑甲酸乙酯固体粗品101g,粗品纯度93.5%,异构体5-二氟甲基吡唑甲酸乙酯含量为6.5%。向粗品中加入正己烷100g,加热回流至全溶,搅拌冷却至室温,结晶、过滤,得到3-二氟甲基吡唑甲酸乙酯固体粗品87g,纯度为99.8%。
[0050]
s3.水解步骤:将87g(0.427mol)3-二氟甲基吡唑甲酸乙酯固体粗品溶入240g甲苯中,滴加10%氢氧化纳水溶液187g(0.469mol),50℃搅拌反应5h,分液,水层加入浓盐酸49g,调节ph值为2~3,大量白色固体析出,过滤,冷水洗涤滤饼,烘干得到产品73g,纯度99.5%,经计算,基于3-(n,n-二甲胺基)丙烯酸乙酯的产品收率为82.9%。
[0051]
实施例2
[0052]
本实施例的操作同实施例1,区别仅在于:
[0053]
在酰化步骤中加入50.5g(0.5mol)三乙胺作为缚酸剂,反应液析出大量糊状固体,
在0℃下滴加水50g,快速搅拌,分液,有机层用50g水洗一次,分液,得到黄色液体作为酰化反应液用于环化步骤。
[0054]
在环化步骤中,得到3-二氟甲基吡唑甲酸乙酯固体粗品99g,粗品纯度87%,异构体5-二氟甲基吡唑甲酸乙酯含量为12%。重结晶后获得3-二氟甲基吡唑甲酸乙酯固体粗品80g,纯度为99.7%。
[0055]
经水解步骤得到产品67.6g,纯度99.5%,经计算,基于3-(n,n-二甲胺基)丙烯酸乙酯的产品收率为76.8%。
[0056]
实施例3
[0057]
本实施例的操作同实施例1,区别仅在于:在水解步骤中,在87g(0.427mol)3-二氟甲基吡唑甲酸乙酯固体中,直接滴加10%氢氧化纳水溶液187g(0.469mol),50℃搅拌反应3h,分液,水层滴加入浓盐酸49g,调节ph值为2~3,大量白色固体析出,过滤,冷水洗涤滤饼,烘干得到产品74.8g,纯度99.4%,经计算,基于3-(n,n-二甲胺基)丙烯酸乙酯的产品收率为85%。
[0058]
实施例4
[0059]
本实施例提供3-(三氟甲基)-1-甲基-1h-吡唑-4-羧酸的制备方法,具体步骤如下:
[0060][0061]
s1.酰化步骤:将71.5g(0.5mol)3-(n,n-二甲胺基)丙烯酸乙酯加入到1l三口瓶中,加入甲苯214.5g,开启搅拌,冷却降温至-5℃,向三口瓶中滴加110.2g(0.525mol)三氟乙酸酐,滴加过程中控制温度在-5~0℃之间,滴加完毕后于0℃下保温反应1h,获得酰化反应液;
[0062]
s2.环化步骤:将60.4g(0.525mol)40%的甲基肼水溶液和71g甲苯混合加入1l三口瓶中,搅拌冷却降温到-10℃,缓慢滴加酰化反应液,控制温度-10℃,滴完保温搅拌1小时,之后升温到室温反应1小时。
[0063]
反应液静置分液,有机层水洗一次后减压蒸除甲苯,得到3-三氟甲基吡唑甲酸乙酯固体粗品105g,粗品纯度92.3%,异构体5-三氟甲基吡唑甲酸乙酯含量为7.5%。向粗品中加入正己烷100g,加热回流至全溶,搅拌冷却至室温,结晶、过滤,得到3-三氟甲基吡唑甲酸乙酯固体粗品93.2g,纯度为99.6%。
[0064]
s3.水解步骤:将93.2g(0.42mol)3-三氟甲基吡唑甲酸乙酯固体粗品溶入240g甲苯中,滴加10%氢氧化纳水溶液187g(0.469mol),50℃搅拌反应5h,分液,水层加入浓盐酸49g,调节ph值为2~3,大量白色固体析出,过滤,冷水洗涤滤饼,烘干得到产品78.1g,纯度99.5%,经计算,基于3-(n,n-二甲胺基)丙烯酸乙酯的产品收率为80.5%。
[0065]
实施例5
[0066]
本实施例的操作同实施例4,区别仅在于:在水解步骤中,在93.2g(0.42mol)3-三氟甲基吡唑甲酸乙酯固体中,直接滴加10%氢氧化纳水溶液187g(0.469mol),50℃搅拌反应3h,分液,水层滴加入浓盐酸49g,调节ph值为2~3,大量白色固体析出,过滤,冷水洗涤滤
h-吡唑甲酸乙酯固体粗品116g,粗品纯度91.8%,异构体5-三氟甲基吡唑甲酸乙酯含量为8.2%。向粗品中加入正己烷116g,加热回流至全溶,搅拌冷却至室温,结晶、过滤,得到3-三氟甲基-1-乙基-1-h-吡唑甲酸乙酯固体粗品99g,纯度为99.6%。
[0080]
s3.水解步骤:将99g(0.419mol)3-三氟甲基-1-乙基-1-h-吡唑甲酸乙酯固体直接滴加10%氢氧化纳水溶液182g(0.462mol),50℃搅拌反应3h,分液,水层滴加入浓盐酸49g,调节ph值为2~3,大量白色固体析出,过滤,冷水洗涤滤饼,烘干得到产品84.5g,纯度99.3%,经计算,基于3-(n,n-二甲胺基)丙烯酸乙酯的产品收率为81.3%。
[0081]
对比例1
[0082]
本对比例提供3-(二氟甲基)-1-甲基-1h-吡唑-4-羧酸的制备方法,采用二氟乙酰氟作为氟化试剂,具体包括以下步骤:
[0083]
(1)酰化步骤:将71.5g(0.5mol)3-(n,n-二甲胺基)丙烯酸乙酯加入到1l三口瓶中,加入甲苯214.5g,三乙胺50.5g(0.5mol),开启搅拌,冷却降温至-5℃,向三口瓶中滴加63g(0.55mol)二氟乙酰氟,过程中控制温度在-5~0℃之间,滴加完毕后于0℃下保温反应2h,反应结束后在0℃下滴加入水50g,快速搅拌,分液,有机层用50g水洗一次,分液,得到黄色液体作为酰化反应液用于环化步骤;
[0084]
(2)环化步骤的操作同实施例3,得到3-二氟甲基吡唑甲酸乙酯固体粗品100g,粗品纯度88%,异构体5-二氟甲基吡唑甲酸乙酯含量为11.8%;重结晶后获得3-二氟甲基吡唑甲酸乙酯固体粗品81g,纯度为99.5%;
[0085]
(3)水解步骤的操作同实施3,得到产品68.5g,纯度99.5%,经计算,基于3-(n,n-二甲胺基)丙烯酸乙酯的产品收率为77.8%。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
