发光元件、发光装置、电子设备、照明装置及新型有机化合物
1.本技术是根据申请日为2012年8月21日、申请号为201280001907.9、发明名称为“发光元件、发光装置、电子设备、照明装置及新型有机化合物”的发明专利申请所提交的分案申请(申请日为2012年8月21日、申请号为201710429638.0、发明名称为“发光元件、发光装置、电子设备、照明装置及新型有机化合物”)的进一步的分案申请。
技术领域
2.本发明涉及一种发光元件、发光装置、电子设备、照明装置及新型有机化合物。
背景技术:
3.近年来,对利用电致发光(el:electro luminescence)的发光元件的研究开发日益火热。在这些发光元件的基本结构中,在一对电极之间夹有包含发光材料的层。通过对该元件施加电压,可以获得来自发光物质的发光。
4.因为这种发光元件是自发光型发光元件,所以具有如下优点:像素的可见度高于液晶显示器;不需要背光灯等。由此,这种发光元件可以被认为适合于平板显示器元件。此外,这种发光元件的重要优点是能够制造成薄型且轻量。再者,非常高速的响应也是这种发光元件的特征之一。
5.进而,由于这种自发光型的发光元件可以形成为膜状,所以能够容易获得面发光,从而能够形成利用面发光的大面积元件。这是在以白炽灯和led为代表的点光源或以荧光灯为代表的线光源中难以得到的特征,因此作为可以应用于照明等的面光源的利用价值也高。
6.根据发光物质是有机化合物还是无机化合物,对上述利用电致发光的发光元件进行大致的分类。在使用将有机化合物用于发光物质且在一对电极之间设置包含该有机化合物的层的有机el元件时,通过对发光元件施加电压,电子和空穴分别从阴极和阳极注入到包含发光有机化合物的层,而使电流流过。而且,所注入的电子及空穴使有机化合物成为激发态,而从所激发的有机化合物得到发光。
7.由有机化合物形成的激发态可以是单重态激发或三重态激发,来自单重态激发(s
*
)的发光被称为荧光,而来自三重态激发(t
*
)的发光被称为磷光。另外,在发光元件中,单重态激发和三重态激发的统计学上的生成比率被认为是s
*
:t
*
=1:3。
8.在将单重态激发能转换成发光的化合物(以下称为荧光化合物)中,在室温下仅观察到来自单重态激发的发光(荧光),观察不到来自三重态激发的发光(磷光)。因此,基于s
*
:t
*
=1:3的关系,使用荧光化合物的发光元件中的内部量子效率(所产生的光子相对于所注入的载流子的比率)的理论上的极限被认为是25%。
9.另一方面,在将三重态激发能转换成发光的化合物(以下称为磷光化合物)中,观察到来自三重态激发的发光(磷光)。此外,在磷光化合物中,由于容易出现系间穿越(即从单重态激发转移到三重态激发),因此理论上内部量子效率能够增加到100%。换句话说,可以得到比荧光化合物高的发射效率。由于该原因,为了实现高效率的发光元件,近年来已在
对使用磷光化合物的发光元件进行深入研究开发。
10.当使用上述磷光化合物形成发光元件的发光层时,为了抑制磷光化合物的浓度猝灭或由三重态-三重态湮灭导致的猝灭,通常以使该磷光化合物分散在由其他化合物构成的基体中的方式形成发光层。此时,用作基体的化合物被称为主体材料,分散在基体中的化合物诸如磷光化合物被称为客体材料(掺杂物)。
11.当将磷光化合物用作客体材料时,主体材料所需要的性质是具有比该磷光化合物高的三重态激发能(基态与三重态激发之间的能量差)。
12.另外,由于单重态激发能(基态与单重态激发之间的能量差)高于三重态激发能,所以具有高三重态激发能的物质也具有高单重态激发能。因此,上述具有高三重态激发能的物质还对将荧光化合物用作发光物质的发光元件有效。
13.作为当磷光化合物为客体材料时使用的主体材料或电子传输材料,对以嘧啶等为部分结构的化合物已经进行了研究(例如专利文献1)。
14.另外,公开了一种化合物,在该化合物中将磷光化合物用作客体材料时,作为主体材料使用组合咔唑骨架与含氮芳香杂环的化合物(例如专利文献2)。
15.[专利文献1]日本专利申请公开第2003-45662号公报
[0016]
[专利文献2]国际专利申请公开第2011-046182号公报
[0017]
如专利文献1或专利文献2所报告,对磷光化合物的主体材料及磷光化合物的客体材料积极地进行研究。但是,从作为发光元件的角度来看,在发光效率、可靠性、发光特性、合成效率或成本等的方面上还有改善的余地,因此需要研发更优越的发光元件。
技术实现要素:
[0018]
鉴于上述问题,本发明的一个方式的目的之一是提供一种发光元件,该发光元件包括用于发光层的发光物质及用作使发光物质分散的主体材料的新颖有机化合物。尤其是,本发明的一个方式的目的之一是提供一种有机化合物,该有机化合物是在作为发光物质使用磷光铱金属配合物的情况下适用于主体材料的新颖有机化合物。
[0019]
另外,本发明的一个方式的目的之一是提供一种包括上述发光元件的发光装置、电子设备及照明装置。
[0020]
本发明的一个方式是一种包括一对电极之间的el层的发光元件。该el层包括第一化合物及第二化合物。第一化合物是lumo(最低未占据分子轨道)能级为-3.5ev以上且-2.5ev以下的磷光铱金属配合物。第二化合物是包含嘧啶骨架的有机化合物。
[0021]
本发明的另一个方式是一种包括一对电极之间的el层的发光元件。该el层包括第一化合物及第二化合物。第一化合物是包含二嗪骨架的磷光铱金属配合物。第二化合物是包含嘧啶骨架的有机化合物。
[0022]
本发明的另一个方式是一种包括一对电极之间的el层的发光元件。该el层包括第一化合物及第二化合物。第一化合物是lumo能级为-3.5ev以上且-2.5ev以下的包含二嗪骨架的磷光铱金属配合物。第二化合物是包含嘧啶骨架的有机化合物。
[0023]
本发明的另一个方式是一种包括一对电极之间的多个el层的发光元件。该多个el层中的至少一层包括第一化合物及第二化合物。第一化合物是lumo能级为-3.5ev以上且-2.5ev以下的包含二嗪骨架的磷光铱金属配合物。第二化合物是包含嘧啶骨架的有机化合
物。
[0024]
在上述各结构中,优选的是,二嗪骨架与铱形成配位键。另外,二嗪骨架优选为嘧啶骨架。
[0025]
在包括第一化合物的客体材料及第二化合物的主体材料的发光元件中,当客体材料及主体材料都具有同种的嘧啶骨架时,载流子从主体材料有效地转移到客体材料。
[0026]
另外,由于本发明的一个方式的第二化合物是杂环化合物,其电子传输性高,所以不仅可以用作发光元件的el层,还可以用作电子传输层或电子注入层等。
[0027]
另外,在一对电极之间设置多个el层的情况下,通过在el层与另一el层之间配置电荷产生层,能够在保持低电流密度的状态下实现高亮度区域中的发光。因为能够保持低电流密度,所以能够实现使用寿命长的元件。
[0028]
另外,在上述各结构中,第一化合物优选是homo(最高占据分子轨道)能级为-6.0ev以上且-5.0ev以下的磷光铱金属配合物。通过采用这种结构,磷光铱金属配合物容易捕获空穴,所以发光元件的空穴迁移率的随时间的变化被抑制。由此,能够期待元件的长使用寿命化。
[0029]
另外,在上述各结构中,第二化合物的分子量优选为2000以下。例如,在利用蒸镀装置蒸镀第二化合物的情况下,如果分子量为2000以下(更优选为1000以下),则能够提高蒸镀效率。另外,为了使所形成的膜的膜性质稳定,优选将其分子量设定为玻璃化转变温度(tg)相当高的500以上。
[0030]
另外,在上述各结构中,第二化合物优选作为取代基至少包含苯骨架、联苯骨架、萘骨架、咔唑骨架、菲骨架、三亚苯骨架、二苯并噻吩骨架和二苯并呋喃骨架中的任一个。当第二化合物包含上述取代基时,具有高磷光能级(也称为三重态能级)。
[0031]
另外,在上述各结构中,第二化合物可以由通式(g1)表示。
[0032][0033]
在通式(g1)中,r1及r2分别独立表示氢、碳数为1至4的烷基、取代或未取代的苯基和取代或未取代的联苯基中的任一个。另外,ar1作为取代基至少包含萘骨架、菲骨架和三亚苯骨架中的任一个。另外,ar2作为取代基至少包含氢、萘骨架、菲骨架和三亚苯骨架中的任一个。
[0034]
另外,在上述各结构中,第二化合物可以由通式(g2)表示。另外,由以下通式(g2)表示的化合物是适用于第二化合物的新颖化合物,该化合物是本发明的一个方式。
[0035][0036]
在通式(g2)中,r1至r5分别独立表示氢、碳数为1至4的烷基、取代或未取代的苯基
和取代或未取代的联苯基中的任一个。另外,ar3表示取代或未取代的萘基、取代或未取代的菲基和取代或未取代的三亚苯基中的任一个。另外,ar4表示氢、取代或未取代的苯基、取代或未取代的联苯基、取代或未取代的萘基、取代或未取代的菲基和取代或未取代的三亚苯基中的任一个。另外,ar5表示氢、碳数为1至4的烷基、取代或未取代的苯基和取代或未取代的联苯基中的任一个。另外,α3及α4分别独立表示取代或未取代的亚苯基。另外,j及k分别独立表示0或1。
[0037]
另外,在上述各结构中,第二化合物可以由通式(g2-1)表示。另外,由以下通式(g2-1)表示的化合物是适用于第二化合物的新颖化合物,该化合物是本发明的一个方式。
[0038][0039]
在通式(g2-1)中,r1至r8分别独立表示氢、碳数为1至4的烷基、取代或未取代的苯基和取代或未取代的联苯基中的任一个。另外,ar3表示取代或未取代的萘基、取代或未取代的菲基和取代或未取代的三亚苯基中的任一个。另外,ar4、ar6及ar7分别独立表示氢、取代或未取代的苯基、取代或未取代的联苯基、取代或未取代的萘基、取代或未取代的菲基和取代或未取代的三亚苯基中的任一个。另外,α3、α4、α6及α7分别独立表示取代或未取代的亚苯基。另外,j、k、m及n分别独立表示0或1。
[0040]
另外,在上述各结构中,第二化合物可以由通式(g3)表示。另外,由以下通式(g3)表示的化合物是适用于第二化合物的新颖化合物,该化合物是本发明的一个方式。
[0041][0042]
在通式(g3)中,r1至r
10
分别独立表示氢、碳数为1至4的烷基、取代或未取代的苯基和取代或未取代的联苯基中的任一个。另外,ar3表示取代或未取代的萘基、取代或未取代的菲基和取代或未取代的三亚苯基中的任一个。另外,ar7表示氢、取代或未取代的苯基、取代或未取代的联苯基、取代或未取代的萘基、取代或未取代的菲基和取代或未取代的三亚苯基中的任一个。另外,α3及α7分别独立表示取代或未取代的亚苯基。另外,j及n分别独立表示0或1。
[0043]
另外,在上述各结构中,第二化合物可以由结构式(300)表示。
[0044][0045]
另外,在上述各结构中,第二化合物的重量比率优选大于第一化合物。换言之,在发光元件中,第二化合物用作主体材料,第一化合物用作客体材料。
[0046]
另外,本发明的另一个方式是由通式(g4)表示的有机化合物。
[0047][0048]
在通式(g4)中,ar
11
、ar
12
、r
11
至r
13
及r
21
至r
23
分别独立表示氢、碳数为1至4的烷基、取代或未取代的苯基和取代或未取代的联苯基中的任一个。另外,ar
13
及ar
14
分别独立表示氢、碳数为1至4的烷基、取代或未取代的苯基、取代或未取代的联苯基、取代或未取代的二苯并噻吩-4-基和取代或未取代的二苯并呋喃-4-基中的任一个。另外,α1、α2、α8及α9分别独立表示取代或未取代的亚苯基。另外,h、i、x及y分别独立表示0或1。另外,e1及e2分别独立表示硫或氧。
[0049]
另外,本发明的另一个方式是由通式(g5)表示的有机化合物。
[0050][0051]
在通式(g5)中,ar
11
、ar
12
、r
11
至r
13
及r
21
至r
23
分别独立表示氢、碳数为1至4的烷基、取代或未取代的苯基和取代或未取代的联苯基中的任一个。另外,ar
13
及ar
14
分别独立表示氢、碳数为1至4的烷基、取代或未取代的苯基、取代或未取代的联苯基、取代或未取代的二苯并噻吩-4-基和取代或未取代的二苯并呋喃-4-基中的任一个。另外,e1及e2分别独立表示硫或氧。
[0052]
另外,本发明的另一个方式是由通式(g6)表示的有机化合物。
[0053][0054]
在通式(g6)中,r
11
至r
13
及r
21
至r
23
分别独立表示氢、碳数为1至4的烷基、取代或未取代的苯基和取代或未取代的联苯基中的任一个。另外,e1及e2分别独立表示硫或氧。
[0055]
另外,本发明的另一个方式是由通式(g7)表示的有机化合物。
[0056][0057]
在通式(g7)中,r
11
至r
13
及r
21
至r
23
分别独立表示氢、碳数为1至4的烷基、取代或未取代的苯基和取代或未取代的联苯基中的任一个。另外,e1及e2分别独立表示硫或氧。
[0058]
另外,本发明的另一个方式是由结构式(400)表示的有机化合物。
[0059][0060]
另外,本发明的一个方式是在一对电极之间包括上述有机化合物的发光元件。尤其是,该发光元件优选在发光层中包括上述有机化合物。
[0061]
另外,本发明包括使用上述发光元件的发光装置、电子设备以及照明装置。另外,本说明书中的发光装置包括图像显示装置、发光装置以及光源。另外,如下模块也都包括在发光装置中:在面板上安装有连接器如fpc(flexible printed circuit,柔性印刷电路)、tab(tape automated bonding,带式自动接合)胶带或tcp(tape carrier package,带载封装)的模块;tab胶带及tcp的端部设置有印刷布线板的模块;通过cog(chip on glass,玻璃覆晶封装)方式将ic(集成电路)直接安装在发光元件中的模块。
[0062]
本发明的一个方式能够提供一种发光元件,该发光元件包括用于发光层的发光物质及用作使发光物质分散的主体材料的新颖有机化合物。尤其是,本发明的一个方式能够提供一种有机化合物,该有机化合物是在作为发光物质使用磷光铱金属配合物的情况下适
用于主体材料的新颖有机化合物。另外,本发明的一个方式能够提供一种驱动电压低且电流效率高的发光元件。根据本发明的一个方式,通过使用上述发光元件,能够提供一种耗电量低的发光装置、电子设备及照明装置。
附图说明
[0063]
图1是说明本发明的一个方式的发光元件的图;
[0064]
图2a和图2b是说明本发明的一个方式的发光元件的图;
[0065]
图3是说明本发明的一个方式的发光元件的图;
[0066]
图4a和图4b是说明本发明的一个方式的发光元件的图;
[0067]
图5是说明本发明的一个方式的发光元件的图;
[0068]
图6a和图6b是说明本发明的一个方式的发光装置的图;
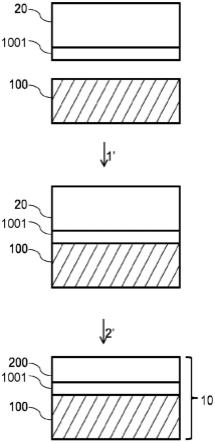
[0069]
图7a和图7b是说明本发明的一个方式的发光装置的图;
[0070]
图8a至图8d是说明本发明的一个方式的电子设备的图;
[0071]
图9a至图9c是说明本发明的一个方式的照明设备的图;
[0072]
图10a和图10b示出4,6mpnp2pm的1h nmr图;
[0073]
图11a和图11b是示出4,6mpnp2pm的甲苯溶液的吸收光谱及发射光谱的图;
[0074]
图12a和图12b是示出4,6mpnp2pm的薄膜的吸收光谱及发射光谱的图;
[0075]
图13a和图13b示出2ph-4,6mnp2pm的1h nmr图;
[0076]
图14a和图14b是示出2ph-4,6mnp2pm的甲苯溶液的吸收光谱及发射光谱的图;
[0077]
图15a和图15b是示出2ph-4,6mnp2pm的薄膜的吸收光谱及发射光谱的图;
[0078]
图16a和图16b示出4,6mtpp2pm的1h nmr图;
[0079]
图17a和图17b是示出4,6mtpp2pm的甲苯溶液的吸收光谱及发射光谱的图;
[0080]
图18a和图18b是示出4,6mtpp2pm的薄膜的吸收光谱及发射光谱的图;
[0081]
图19a和图19b示出4,6mdbtp2pm-ii的1h nmr图;
[0082]
图20a和图20b是示出4,6mdbtp2pm-ii的甲苯溶液的吸收光谱及发射光谱的图;
[0083]
图21a和图21b是示出4,6mdbtp2pm-ii的薄膜的吸收光谱及发射光谱的图;
[0084]
图22a和图22b示出2,4dbtp2pm-ii的1h nmr图;
[0085]
图23a和图23b是示出2,4dbtp2pm-ii的甲苯溶液的吸收光谱及发射光谱的图;
[0086]
图24a和图24b是示出2,4dbtp2pm-ii的薄膜的吸收光谱及发射光谱的图;
[0087]
图25a和图25b示出2,5dbtp2pm-ii的1h nmr图;
[0088]
图26a和图26b是示出2,5dbtp2pm-ii的甲苯溶液的吸收光谱及发射光谱的图;
[0089]
图27a和图27b是示出2,5dbtp2pm-ii的薄膜的吸收光谱及发射光谱的图;
[0090]
图28a和图28b示出4,6mdbtp2pm-iii的1h nmr图;
[0091]
图29a和图29b是示出4,6mdbtp2pm-iii的甲苯溶液的吸收光谱及发射光谱的图;
[0092]
图30a和图30b是示出4,6mdbtp2pm-iii的薄膜的吸收光谱及发射光谱的图;
[0093]
图31a和图31b示出4,6mdbfp2pm-ii的1h nmr图;
[0094]
图32a和图32b是示出4,6mdbfp2pm-ii的甲苯溶液的吸收光谱及发射光谱的图;
[0095]
图33a和图33b是示出4,6mdbfp2pm-ii的薄膜的吸收光谱及发射光谱的图;
[0096]
图34a和图34b示出2,4dbfp2pm-ii的1h nmr图;
[0097]
图35a和图35b是示出2,4dbfp2pm-ii的甲苯溶液的吸收光谱及发射光谱的图;
[0098]
图36a和图36b是示出2,4dbfp2pm-ii的薄膜的吸收光谱及发射光谱的图;
[0099]
图37a和图37b示出2,5dbfp2pm-ii的1h nmr图;
[0100]
图38a和图38b是示出2,5dbfp2pm-ii的甲苯溶液的吸收光谱及发射光谱的图;
[0101]
图39a和图39b是示出2,5dbfp2pm-ii的薄膜的吸收光谱及发射光谱的图;
[0102]
图40是说明实施例的发光元件1的图;
[0103]
图41是示出发光元件1的电流密度-亮度特性的图;
[0104]
图42是示出发光元件1的电压-亮度特性的图;
[0105]
图43是示出发光元件1的亮度-电流效率特性的图;
[0106]
图44是示出发光元件1的电压-电流特性的图;
[0107]
图45是示出发光元件1的亮度-色度坐标特性的图;
[0108]
图46是示出发光元件1的亮度-功率效率特性的图;
[0109]
图47是示出发光元件1的发射光谱的图;
[0110]
图48是示出发光元件1的时间-归一化亮度特性的图;
[0111]
图49是示出发光元件1的时间-电压特性的图;
[0112]
图50a至图50c是说明实施例的发光元件2至7的图;
[0113]
图51是示出发光元件2的电流密度-亮度特性的图;
[0114]
图52是示出发光元件2的电压-亮度特性的图;
[0115]
图53是示出发光元件2的亮度-电流效率特性的图;
[0116]
图54是示出发光元件2的电压-电流特性的图;
[0117]
图55是示出发光元件2的亮度-色度坐标特性的图;
[0118]
图56是示出发光元件2的亮度-功率效率特性的图;
[0119]
图57是示出发光元件2的发射光谱的图;
[0120]
图58是示出发光元件3的电流密度-亮度特性的图;
[0121]
图59是示出发光元件3的电压-亮度特性的图;
[0122]
图60是示出发光元件3的亮度-电流效率特性的图;
[0123]
图61是示出发光元件3的电压-电流特性的图;
[0124]
图62是示出发光元件3的亮度-色度坐标特性的图;
[0125]
图63是示出发光元件3的亮度-功率效率特性的图;
[0126]
图64是示出发光元件3的发射光谱的图;
[0127]
图65是示出发光元件4的电流密度-亮度特性的图;
[0128]
图66是示出发光元件4的电压-亮度特性的图;
[0129]
图67是示出发光元件4的亮度-电流效率特性的图;
[0130]
图68是示出发光元件4的电压-电流特性的图;
[0131]
图69是示出发光元件4的亮度-色度坐标特性的图;
[0132]
图70是示出发光元件4的亮度-功率效率特性的图;
[0133]
图71是示出发光元件4的发射光谱的图;
[0134]
图72是示出发光元件5的电流密度-亮度特性的图;
[0135]
图73是示出发光元件5的电压-亮度特性的图;
[0136]
图74是示出发光元件5的亮度-电流效率特性的图;
[0137]
图75是示出发光元件5的电压-电流特性的图;
[0138]
图76是示出发光元件5的亮度-色度坐标特性的图;
[0139]
图77是示出发光元件5的亮度-功率效率特性的图;
[0140]
图78是示出发光元件5的发射光谱的图;
[0141]
图79是示出发光元件6的电流密度-亮度特性的图;
[0142]
图80是示出发光元件6的电压-亮度特性的图;
[0143]
图81是示出发光元件6的亮度-电流效率特性的图;
[0144]
图82是示出发光元件6的电压-电流特性的图;
[0145]
图83是示出发光元件6的亮度-色度坐标特性的图;
[0146]
图84是示出发光元件6的亮度-功率效率特性的图;
[0147]
图85是示出发光元件6的发射光谱的图;
[0148]
图86是示出发光元件7的电流密度-亮度特性的图;
[0149]
图87是示出发光元件7的电压-亮度特性的图;
[0150]
图88是示出发光元件7的亮度-电流效率特性的图;
[0151]
图89是示出发光元件7的电压-电流特性的图;
[0152]
图90是示出发光元件7的亮度-色度坐标特性的图;
[0153]
图91是示出发光元件7的亮度-功率效率特性的图;
[0154]
图92是示出发光元件7的发射光谱的图;
[0155]
图93是示出发光元件4的时间-归一化亮度特性的图;
[0156]
图94是示出发光元件4的时间-电压特性的图;
[0157]
图95是示出发光元件5的时间-归一化亮度特性的图;
[0158]
图96是示出发光元件5的时间-电压特性的图;
[0159]
图97是示出4,6mpnp2pm的lc-ms测定结果的图;
[0160]
图98a至图98d是示出4,6mpnp2pm的tof-sims测定结果的图;
[0161]
图99是示出2ph-4,6mnp2pm的lc-ms测定结果的图;
[0162]
图100是示出4,6mtpp2pm的lc-ms测定结果的图;
[0163]
图101是示出4,6mdbtp2pm-ii的lc-ms测定结果的图;
[0164]
图102a至图102d是示出4,6mdbtp2pm-ii的tof-sims测定结果的图;
[0165]
图103是示出2,5mdbtp2pm-ii的lc-ms测定结果的图;
[0166]
图104是示出4,6mdbtp2pm-iii的lc-ms测定结果的图;
[0167]
图105是示出4,6mdbfp2pm-ii的lc-ms测定结果的图;
[0168]
图106是示出2,4mdbfp2pm-ii的lc-ms测定结果的图;
[0169]
图107是示出2,5mdbfp2pm-ii的lc-ms测定结果的图。
具体实施方式
[0170]
以下,参照附图详细地说明本发明的实施方式。但是,本发明不局限于以下说明,而所属技术领域的普通技术人员可以很容易地理解一个事实就是其方式及详细内容在不脱离本发明的宗旨及其范围的情况下可以被变换为各种各样的形式。因此,本发明不应该
被解释为仅局限在以下所示的实施方式所记载的内容中。
[0171]
实施方式1
[0172]
在本实施方式中,参照图1对如下发光元件进行说明,该发光元件具有第一化合物的磷光铱金属配合物及第二化合物的包含嘧啶骨架的有机化合物。
[0173]
在本实施方式所示的发光元件中,如图1所示,在一对电极(第一电极101与第二电极103)之间夹有包含发光层113的el层102,el层102除了发光层113之外,还包含空穴注入层111、空穴传输层112、电子传输层114、电子注入层115、电荷产生层116等而形成。注意,在本实施方式的说明中,将第一电极101用作阳极,而将第二电极103用作阴极。另外,第一电极101设置在衬底100上,作为衬底100可以使用玻璃衬底等。
[0174]
通过对这种发光元件施加电压,从第一电极101一侧注入的空穴和从第二电极103一侧注入的电子在发光层113中重新结合以使第一化合物的磷光铱金属配合物成为激发态。并且,当该处于激发态的第一化合物的磷光铱金属配合物回到基态时发光。像这样,在本发明的一个实施方式中,第一化合物的磷光铱金属配合物用作发光元件中的发光物质。
[0175]
另外,el层102中的空穴注入层111是包含空穴传输性高的物质和受主物质的层,由于受主物质,从空穴传输性高的物质抽出电子,由此产生空穴。因此,空穴从空穴注入层111经过空穴传输层112注入到发光层113。
[0176]
另外,电荷产生层116是包含空穴传输性高的物质和受主物质的层。由于受主物质,从空穴传输性高的物质抽出电子,因此被抽出的电子从具有电子注入性的电子注入层115经过电子传输层114注入到发光层113。
[0177]
下面,对制造本实施方式所示的发光元件时的具体例子进行说明。
[0178]
作为第一电极101及第二电极103,可以使用金属、合金、导电化合物及它们的混合物等。具体而言,除了氧化铟-氧化锡(ito:indium tin oxide)、包含硅或氧化硅的氧化铟-氧化锡、氧化铟-氧化锌(氧化铟锌)、包含氧化钨及氧化锌的氧化铟、金(au)、铂(pt)、镍(ni)、钨(w)、铬(cr)、钼(mo)、铁(fe)、钴(co)、铜(cu)、钯(pd)、钛(ti)之外,还可以使用属于元素周期表中第1族或第2族的元素,即碱金属诸如锂(li)和铯(cs)等、碱土金属诸如钙(ca)和锶(sr)等、镁(mg)、包含它们的合金(mgag、alli)、稀土金属诸如铕(eu)和镱(yb)等、包含它们的合金及石墨烯等。另外,第一电极101及第二电极103可以通过溅射法或蒸镀法(包括真空蒸镀法)等形成。
[0179]
作为用于空穴注入层111、空穴传输层112及电荷产生层116的空穴传输性高的物质,例如可以举出4,4
′‑
双[n-(1-萘基)-n-苯基氨基]联苯(简称:npb或α-npd)、n,n
′‑
双(3-甲基苯基)-n,n
′‑
二苯基-[1,1
′‑
联苯]-4,4
′‑
二胺(简称:tpd)、4,4
′
,4
″‑
三(咔唑-9-基)三苯胺(简称:tcta)、4,4
′
,4
″‑
三(n,n-二苯基氨基)三苯胺(简称:tdata)、4,4
′
,4
″‑
三[n-(3-甲基苯基)-n-苯基氨基]三苯胺(简称:mtdata)、4,4
′‑
双[n-(螺-9,9
′‑
联芴-2-基)-n-苯基氨基]联苯(简称:bspb)等芳香胺化合物;3-[n-(9-苯基咔唑-3-基)-n-苯基氨基]-9-苯基咔唑(简称:pczpca1)、3,6-双[n-(9-苯基咔唑-3-基)-n-苯基氨基]-9-苯基咔唑(简称:pczpca2)、3-[n-(1-萘基)-n-(9-苯基咔唑-3-基)氨基]-9-苯基咔唑(简称:pczpcn1)等。除上述以外,还可以使用:4,4
′‑
二(n-咔唑基)联苯(简称:cbp)、1,3,5-三[4-(n-咔唑基)苯基]苯(简称:tcpb)、9-[4-(10-苯基-9-蒽基)苯基]-9h-咔唑(简称:czpa)等的咔唑化合物;1,3,5-三(二苯并噻吩-4-基)-苯(简称:dbt3p-ii)等二苯并噻吩化合物;1,3,5-三(二苯并
呋喃-4-基)苯(简称:dbf3p-ii)等二苯并呋喃化合物;9-[3,5-二-(菲-9-基)-苯基]-菲(简称:pn3p)等稠环化合物等。这些物质主要是各自具有10-6
cm2/vs以上的空穴迁移率的物质。注意,还可以使用除上述材料之外的物质,只要该物质的空穴传输性高于电子传输性。
[0180]
再者,还可以使用聚(n-乙烯基咔唑)(简称:pvk)、聚(4-乙烯三苯胺)(简称:pvtpa)、聚[n-(4-{n
′‑
[4-(4-二苯基氨基)苯基]苯基-n
′‑
苯基氨基}苯基)甲基丙烯酰胺](简称:ptpdma)、聚[n,n
′‑
双(4-丁基苯基)-n,n
′‑
双(苯基)联苯胺](简称:poly-tpd)等高分子化合物。
[0181]
另外,空穴注入层111及电荷产生层116可以使用含有上述空穴传输性高的物质和受主物质的层。在此情况下,载流子注入性得到提高,所以是优选的。作为用于空穴注入层111及电荷产生层116的受主物质,可以举出过渡金属氧化物或属于元素周期表中第4族至第8族的金属的氧化物。具体地说,氧化钼是特别优选的。
[0182]
发光层113作为用作发光物质的客体材料包含第一化合物的磷光铱金属配合物,并且作为主体材料包含其三重态激发能高于该第一化合物的磷光铱金属配合物的物质。
[0183]
在此,作为上述客体材料使用作为第一化合物且lumo能级为-3.5ev以上且-2.5ev以下的磷光铱金属配合物。另外,作为上述主体材料使用作为第二化合物的包含嘧啶骨架的有机化合物。
[0184]
由于包含嘧啶骨架的有机化合物的lumo能级受到嘧啶骨架的影响(lumo轨道存在于嘧啶骨架附近),所以该化合物的lumo能级位于-3.0ev左右。从而,通过作为第一有机化合物与第二有机化合物的组合采用上述化合物,载流子(电子)从主体材料有效地转移到客体材料,由此客体材料容易有效地进行发光,而且使用寿命也得到提高。同时,客体材料不容易捕获电子,所以不妨碍来源于主体材料的嘧啶骨架的良好的电子传输性,而能够实现元件的低电压化。另外,从这种观点来看,主体材料的lumo能级也优选为-3.5ev以上且-2.5ev以下。
[0185]
另外,第二化合物的包含嘧啶骨架的有机化合物优选作为取代基至少包含苯骨架、联苯骨架、萘骨架、咔唑骨架、菲骨架、三亚苯骨架、二苯并噻吩骨架和二苯并呋喃骨架中的任一个。通过采用这种结构,第二化合物的lumo能级受到嘧啶骨架的很大影响(lumo轨道存在于嘧啶骨架附近),所以上述效果变得更明显。
[0186]
尤其是,当第二化合物的包含嘧啶骨架的有机化合物包含咔唑骨架时,还能够容易转移空穴,由此得到双极性,所以是优选的。当第二化合物的包含嘧啶骨架的有机化合物包含萘骨架、菲骨架、三亚苯骨架等稠环时,载流子传输性得到提高,所以是优选的。当第二化合物的包含嘧啶骨架的有机化合物包含二苯并噻吩骨架或二苯并呋喃骨架时,能够获得立体结构及稳定的膜性质,所以是优选的(尤其是,这些二苯并噻吩骨架和二苯并呋喃骨架的4位取代产物具有电化学稳定性,所以是优选的)。
[0187]
换言之,第二化合物是由通式(g1)表示的有机化合物。
[0188][0189]
在通式(g1)中,r1及r2分别独立表示氢、碳数为1至4的烷基、取代或未取代的苯基
和取代或未取代的联苯基中的任一个。另外,ar1作为取代基至少包含萘骨架、菲骨架和三亚苯骨架中的任一个。另外,ar2作为取代基至少包含氢、萘骨架、菲骨架和三亚苯骨架中的任一个。
[0190]
在此,当ar1与ar2是同样的取代基时易于合成,所以是优选的。另一方面,当ar1与ar2是不同的取代基时获得更立体的结构,所以是优选的。
[0191]
另外,由上述通式(g1)表示的第二化合物更优选具有由通式(g2)表示的结构。另外,由通式(g2)表示的化合物是适用于第二化合物的新型化合物,该化合物是本发明的一个实施方式。
[0192][0193]
在通式(g2)中,r1至r5分别独立表示氢、碳数为1至4的烷基、取代或未取代的苯基和取代或未取代的联苯基中的任一个。另外,ar3表示取代或未取代的萘基、取代或未取代的菲基和取代或未取代的三亚苯基中的任一个。另外,ar4表示氢、取代或未取代的苯基、取代或未取代的联苯基、取代或未取代的萘基、取代或未取代的菲基和取代或未取代的三亚苯基中的任一个。另外,ar5表示氢、碳数为1至4的烷基、取代或未取代的苯基和取代或未取代的联苯基中的任一个。另外,α3及α4分别独立表示取代或未取代的亚苯基。另外,j及k分别独立表示0或1。
[0194]
具体地说,由上述通式(g1)、(g2)表示的第二化合物更优选具有由通式(g2-1)表示的结构。另外,由通式(g2-1)表示的化合物是适用于第二化合物的新型化合物,该化合物是本发明的一个实施方式。
[0195][0196]
在通式(g2-1)中,r1至r8分别独立表示氢、碳数为1至4的烷基、取代或未取代的苯基和取代或未取代的联苯基中的任一个。另外,ar3表示取代或未取代的萘基、取代或未取代的菲基和取代或未取代的三亚苯基中的任一个。另外,ar4、ar6及ar7分别独立表示氢、取代或未取代的苯基、取代或未取代的联苯基、取代或未取代的萘基、取代或未取代的菲基和取代或未取代的三亚苯基中的任一个。另外,α3、α4、α6及α7分别独立表示取代或未取代的亚苯基。另外,j、k、m及n分别独立表示0或1。
[0197]
具体地说,由上述通式(g1)、(g2)及(g2-1)表示的第二化合物更优选具有由通式(g3)表示的结构。另外,由通式(g3)表示的化合物是适用于第二化合物的新型化合物,该化
合物是本发明的一个实施方式。
[0198][0199]
在通式(g3)中,r1至r
10
分别独立表示氢、碳数为1至4的烷基、取代或未取代的苯基和取代或未取代的联苯基中的任一个。另外,ar3表示取代或未取代的萘基、取代或未取代的菲基和取代或未取代的三亚苯基中的任一个。另外,ar7表示氢、取代或未取代的苯基、取代或未取代的联苯基、取代或未取代的萘基、取代或未取代的菲基和取代或未取代的三亚苯基中的任一个。另外,α3及α7分别独立表示取代或未取代的亚苯基。另外,j及n分别独立表示0或1。
[0200]
当上述通式(g2)、(g2-1)及(g3)中的ar1至ar7具有取代基时,该取代基选自碳数为1至4的烷基、取代或未取代的苯基和取代或未取代的联苯基中的任一个。当选择烷基时,在有机溶剂中的溶解性得到提高,易于合成及利用湿法的成膜,所以是优选的。另外,当选择取代或未取代的苯基或取代或未取代的联苯基时,载流子传输性得到提高,所以是优选的。另外,当包含这些取代基时,能够获得更立体的结构且膜性质稳定,所以是优选的。然而,如果因这些取代基而增加合成步骤,则优选不具有取代基。
[0201]
当上述通式(g1)、(g2-1)及(g3)中的r1和r2的双方或者通式(g2)中的r1、r2和ar5中的任两个为氢时,易于合成,所以是优选的。另外,当选择氢时,载流子(电子)注入性得到提高,能够期待低电压化,所以是优选的。当上述通式(g1)、(g2-1)及(g3)中的r1和r2的双方或者通式(g2)中的r1、r2和ar5中的任两个分别独立为碳数为1至4的烷基、取代或未取代的苯基或取代或未取代的联苯基时,非晶性得到提高,膜性质稳定,所以是优选的。
[0202]
在上述通式(g2)、(g2-1)及(g3)中,芳基(ar3至ar7)通过苯骨架键合到嘧啶的4位或6位。这是为了防止共轭从嘧啶骨架扩展到芳基的缘故。再者,因为这些芳基键合于该苯骨架的间位,所以共轭更不容易扩展,homo能级变深。因此,homo能级与lumo能级之间的带隙(bg)容易变宽,所以s1能级及t1能级容易提高。因此,该化合物能够用作发射更短波长的光的掺杂物的主体材料,作为主体材料的使用范围扩大,所以是优选的。更具体地说,该化合物适用于在可见光区(蓝色至红色)发射磷光的材料或在可见光区(蓝色至红色)发射荧光的材料的主体材料。另外,该化合物的homo能级深,所以适用于homo能级深的发光材料的主体材料。
[0203]
作为通式(g1)、(g2)、(g2-1)及(g3)中的ar1至ar7的具体结构,例如可以举出由结构式(ar-1)至结构式(ar-5)表示的取代基。
[0204]
[0205]
另外,作为通式(g2)、(g2-1)及(g3)中的α3、α4、α6及α7的具体结构,例如可以举出由结构式(α-1)至结构式(α-3)表示的取代基。
[0206][0207]
当采用如结构式(α-1)那样的键合于对位的取代基的情况下,载流子传输性得到提高,所以是优选的。当采用如结构式(α-2)或结构式(α-3)那样的键合于间位或邻位的取代基的情况下,t1能级或s1能级得到提高,所以是优选的。
[0208]
另外,作为由上述通式(g1)、(g2)、(g2-1)及(g3)表示的第二化合物的具体例子,可以举出由如下结构式表示的有机化合物:结构式(100)至结构式(107);结构式(110)至结构式(123);结构式(130)至结构式(135);结构式(140)至结构式(145);结构式(150)至结构式(161);结构式(300)至结构式(321)。另外,其中作为由通式(g2)表示的第二化合物的具体例子,可以举出由如下结构式表示的有机化合物:结构式(306)至结构式(309);结构式(318);结构式(320)。另外,作为由上述通式(g2-1)及(g3)表示的第二化合物的具体例子,可以举出由如下结构式表示的有机化合物:结构式(300)至结构式(305);结构式(310)至结构式(317);结构式(319);结构式(321)。注意,本发明不局限于这些化合物。
[0209]
[0210]
[0211]
[0212]
[0213]
[0214]
[0215]
[0216]
[0217]
[0218]
[0219]
[0220][0221]
另外,作为第二化合物的合成方法可以应用各种反应。例如,通过进行下面描述的合成反应能够合成由通式(g1)表示的第二化合物。另外,第二化合物的合成方法不局限于以下合成方法。
[0222]
《由通式(g1)表示的第二化合物的合成方法》
[0223]
首先,以下示出合成方案(a-1)。如合成方案(a-1)所示,通过将二卤化嘧啶有机化合物(a1)与芳基硼有机化合物(a2)偶联,能够合成卤化嘧啶有机化合物(a3)。
[0224][0225]
另外,在合成方案(a-1)中,x1及x2表示氢或卤素。注意,当x1及x2表示卤素时,从高
反应性的观点来看,x1及x2优选表示溴,更优选表示碘。b1表示硼酸或二烷氧基硼(dialkoxyboron)。另外,r1及r2分别独立表示氢、碳数为1至4的烷基、取代或未取代的苯基和取代或未取代的联苯基中的任一个。另外,ar1作为取代基至少包含取代或未取代的萘基、取代或未取代的菲基和取代或未取代的三亚苯基中的任一个。
[0226]
另外,多种反应条件可以用于合成方案(a-1)中的偶联反应。作为其中一个例子,可以采用在碱的存在下使用金属催化剂的合成方法。
[0227]
下面示出在合成方案(a-1)中采用铃木-宫浦反应的例子。可以使用钯催化剂作为金属催化剂,钯配合物和其配体的混合物可以用作钯催化剂。此外,作为钯配合物的例子,可以举出乙酸钯(ii)、四(三苯基膦)钯(0)和双(三苯基膦)二氯化钯(ii)等。作为配体的例子,可以举出三(邻甲苯基)膦、三苯基膦和三环己基膦等。此外,作为可以用作碱的物质的例子,可以举出有机碱如叔丁醇钠以及无机碱如碳酸钠或碳酸钾等。所述反应优选在溶液中进行。可以使用的溶剂的例子如下所示:乙腈和水的混合溶剂;甲苯或二甲苯等稀释剂类和水的混合溶剂;甲苯或二甲苯、乙醇等醇和水的三种混合溶剂;1,3-二甲基-3,4,5,6-四氢-2(1h)-嘧啶酮(dmpu)和水的混合溶剂;乙二醇二甲基醚等醚类和水的混合溶剂等。但是,可以使用的催化剂、配体、碱、溶剂不局限于这些。此外,在合成方案(a-1)中,可以使用芳基铝化合物、芳基锆化合物、芳基锌化合物或芳基锡化合物等代替芳基硼化合物(a2)。另外,反应优选在氮气、氩气等惰性气氛下进行。另外,也可以利用电磁波进行加热。
[0228]
接着,如以下合成方案(a-2)所示,通过将卤化嘧啶有机化合物(a3)与芳基硼有机化合物(a4)偶联可以合成由通式(g1)表示的第二化合物。
[0229][0230]
另外,x2表示氢或卤素。注意,当x2表示卤素时,从高反应性的观点来看,x2优选表示溴,更优选表示碘。b2表示硼酸或二烷氧基硼。另外,r1及r2分别独立表示氢、碳数为1至4的烷基、取代或未取代的苯基和取代或未取代的联苯基中的任一个。另外,ar1作为取代基至少包含取代或未取代的萘基、取代或未取代的菲基和取代或未取代的三亚苯基中的任一个。
[0231]
另外,多种反应条件可以用于合成方案(a-2)中的偶联反应。作为其中一个例子,可以采用在碱的存在下使用金属催化剂的合成方法。
[0232]
在合成方案(a-2)中可以采用铃木-宫浦反应。详细内容可以参照上述合成方案(a-1)进行合成。
[0233]
另外,当ar1与ar2相同时,可以同时进行上述合成方案(a-1)和(a-2)的反应。换言之,能够一同将(a2)及(a4)加入有机化合物(a1)中发生反应,易于合成,所以是优选的。
[0234]
通过上述步骤可以合成本发明的一个实施方式的第二化合物。
[0235]
另外,当发光层113包含上述第二化合物(主体材料)及第一化合物(客体材料)时,可以从发光层113得到发光效率高的磷光发光。
[0236]
另外,作为第一化合物的具体例子,可以举出由如下结构式表示的磷光铱金属配
合物:结构式(200)至结构式(206);结构式(210)至结构式(213);结构式(220)至结构式(222);结构式(230);结构式(231);结构式(240)。注意,本发明不局限于这些化合物。
[0237]
结构式(200)至结构式(206)表示具有嘧啶骨架的磷光铱金属配合物;结构式(210)至结构式(213)表示具有吡嗪骨架的磷光铱金属配合物;结构式(220)至结构式(222)表示具有吡啶骨架或喹啉骨架的磷光铱金属配合物;结构式(230)及结构式(231)表示具有喹喔啉骨架的磷光铱金属配合物,结构式(240)表示具有三嗪骨架的磷光铱金属配合物。
[0238]
[0239]
[0240][0241]
另外,磷光铱金属配合物的结构不局限于上述结构。由于第二化合物的包含嘧啶骨架的有机化合物具有较高的t1能级,所以能够将其用作发射蓝绿光或比蓝绿色更长的波长的光的磷光发光材料的主体材料。注意,如上所述,磷光铱金属配合物的lumo能级优选为-3.5ev以上且-2.5ev以下。
[0242]
另外,磷光铱金属配合物的homo能级优选为-6.0ev以上且-5.0ev以下。通过采用上述结构,磷光铱金属配合物容易捕获空穴,所以发光元件的空穴迁移率的随时间的变化被抑制。由此,能够期待元件的长使用寿命化。尤其是,当第二化合物的包含嘧啶骨架的有机化合物作为取代基至少包含苯骨架、联苯骨架、萘骨架、菲骨架、三亚苯骨架、二苯并噻吩骨架和二苯并呋喃骨架中的任一个时,第二化合物的包含嘧啶骨架的有机化合物的homo能级为-6.0ev以下,所以上述空穴捕获的效果变得更明显。
[0243]
在此,在本发明的另一个实施方式中,优选的是,主体材料具有嘧啶骨架,客体材料具有二嗪骨架。另外,更优选的是,主体材料及客体材料都具有嘧啶骨架。当采用上述材料时,这些材料之间的lumo能级相近。结果,载流子(电子)从主体材料有效地转移到客体材料,由此客体材料容易有效地进行发光,而且使用寿命也得到提高。另外,当这些材料的lumo能级相近时,客体材料不容易捕获电子,所以不妨碍来源于主体材料的嘧啶骨架的良好的电子传输性,而能够实现元件的低电压化。从而,能够期待发光元件的高效率化、长使用寿命化及低驱动电压化。当主体材料及客体材料都具有嘧啶骨架时,主体分子与客体分子之间的相互作用变强,所以上述效果变得更明显。
[0244]
另外,能够选择具有彼此相近的lumo能级的主体材料和客体材料的原因如下所述。首先,主体材料的lumo能级受到容易被还原的二嗪骨架之一的嘧啶骨架的影响。尤其是,由通式(g1)、(g2)、(g2-1)及(g3)表示的主体材料的lumo轨道存在于嘧啶骨架附近。另一方面,客体材料也具有容易被还原的二嗪骨架,所以客体材料的lumo能级也受到二嗪骨
架的影响。尤其是,当二嗪骨架与铱形成配位键时,lumo轨道不存在于中心金属而存在于二嗪骨架。从而,主体材料与客体材料都具有来源于二嗪骨架的lumo轨道,所以这些材料的lumo能级也相近。
[0245]
另外,当如上所述那样磷光铱金属配合物(第一化合物)具有二嗪骨架尤其具有嘧啶骨架时,该磷光铱金属配合物的lumo能级优选为-3.5ev以上且-2.5ev以下。另外,其homo能级优选为-6.0ev以上且-5.0ev以下。另外,该情况下的主体材料(第二化合物)的优选的例子是上面所示的。
[0246]
另外,如上所述,本发明的一个方式的第二化合物(主体材料)的lumo轨道存在于容易被还原且电子传输性良好的嘧啶骨架附近。从而,本发明的一个方式的第二化合物具有高电子传输性并降低元件的驱动电压。
[0247]
另外,在实施方式1中说明使用第一化合物的磷光铱金属配合物的磷光发光元件,但是不局限于上述结构。第二化合物的包含嘧啶骨架的有机化合物具有高t1能级,所以还具有高s1能级。因此,第二化合物的包含嘧啶骨架的有机化合物还可以用作在可见光区发射荧光的材料的主体材料。
[0248]
另外,可以使用多种用来分散发光物质(客体材料)的物质(主体材料)。因此,除了第二化合物的包含嘧啶骨架的有机化合物以外,发光层还可以包含第二主体材料。
[0249]
作为第二主体材料,例如可以举出用于空穴传输层112的材料。
[0250]
电子传输层114是包含电子传输性高的物质的层。作为电子传输层114,可以使用金属配合物诸如alq3、三(4-甲基-8-羟基喹啉合)铝(简称:almq3)、双(10-羟基苯并[h]喹啉合)铍(简称:bebq2)、balq、zn(box)2或双[2-(2-羟基苯基)苯并噻唑]锌(简称:zn(btz)2)等。此外,也可以使用杂芳族化合物诸如2-(4-联苯基)-5-(4-叔丁基苯基)-1,3,4-二唑(简称:pbd)、1,3-双[5-(对叔丁基苯基)-1,3,4
‑ꢀ
二唑-2-基]苯(简称:oxd-7)、3-(4-叔丁基苯基)-4-苯基-5-(4-联苯基)-1,2,4-三唑(简称:taz)、3-(4-叔丁基苯基)-4-(4-乙基苯基)-5-(4-联苯基)-1,2,4-三唑(简称:p-ettaz)、红菲咯啉(简称:bphen)、浴铜灵(简称:bcp)、4,4
′‑
双(5-甲基苯并 唑-2-基)均二苯乙烯(简称:bzos)等。另外,还可以使用高分子化合物诸如聚(2,5-吡啶二基)(简称:ppy)、聚[(9,9-二己基芴-2,7-二基)-共-(吡啶-3,5-二基)](简称:pf-py)、聚[(9,9-二辛基芴-2,7-二基)-共-(2,2
′‑
联吡啶-6,6
′‑
二基)](简称:pf-bpy)。在此所述的物质主要是电子迁移率为10-6
cm2/vs以上的物质。另外,只要是电子传输性比空穴传输性高的物质,就可以将上述物质之外的物质用于电子传输层。
[0251]
另外,本发明中使用的第二化合物的包含嘧啶骨架的有机化合物也是电子传输性良好的材料,所以该化合物适用于电子传输层。
[0252]
另外,作为电子传输层114,不仅可以采用单层,而且可以采用由上述物质构成的层的两层以上的叠层。
[0253]
电子注入层115是包含电子注入性高的物质的层。作为电子注入层115,可以使用碱金属、碱土金属或氟化锂(lif)、氟化铯(csf)、氟化钙(caf2)及锂氧化物(lio
x
)等它们的化合物。此外,可以使用氟化铒(erf3)等稀土金属化合物。另外,也可以使用上述构成电子传输层114的物质。
[0254]
或者,也可以将有机化合物与电子给体(供体)混合而成的复合材料用于电子注入层115。因为在这种复合材料中电子给体使电子产生在有机化合物中,所以电子注入性及电
子传输性高。在此情况下,有机化合物优选是能够高效地传输所产生的电子的材料。具体地,例如,可以使用如上所述的构成电子传输层114的物质(金属配合物或杂芳族化合物等)。作为电子给体,只要使用对有机化合物呈现电子给体性的物质,即可。具体地,优选使用碱金属、碱土金属和稀土金属,可以举出锂、铯、镁、钙、铒、镱等。另外,优选使用碱金属氧化物或碱土金属氧化物,例如可以举出锂氧化物、钙氧化物、钡氧化物等。此外,可以使用氧化镁等路易斯碱。或者,也可以使用四硫富瓦烯(简称:ttf)等有机化合物。
[0255]
另外,上述空穴注入层111、空穴传输层112、发光层113、电子传输层114、电子注入层115和电荷产生层116分别可以通过蒸镀法(包括真空蒸镀法)、喷墨法、涂敷法等的方法形成。
[0256]
在上述发光元件中,由于在第一电极101与第二电极103之间产生的电位差而产生电流,并且在el层102中空穴和电子重新结合而发光。然后,该发光穿过第一电极101和第二电极103中的任一方或双方取出到外部。因此,第一电极101和第二电极103中的任一方或双方为具有透光性的电极。
[0257]
因为如上所说明的发光元件可以得到来源于第一化合物的磷光铱金属配合物的磷光发光,所以可以实现与使用荧光化合物的发光元件相比效率高的发光元件。
[0258]
另外,本实施方式所示的发光元件是发光元件的结构的一个例子。也可以将其他实施方式所示的结构的发光元件用于本发明的一个实施方式的发光装置。此外,作为具备上述发光元件的发光装置,可以制造无源矩阵型发光装置或有源矩阵型发光装置。此外,还可以制造具备在其他实施方式说明的与上述发光元件不同的发光元件的微腔结构的发光装置等。上述发光装置都包括在本发明中。
[0259]
另外,在有源矩阵型发光装置的情况下,对tft的结构没有特别的限制。例如,可以适当地使用交错型tft或反交错型tft。此外,形成在tft衬底上的驱动电路可以由n型tft和p型tft中的一方或双方形成。并且,对用于tft的半导体膜的结晶性也没有特别的限制。例如,可以使用非晶半导体膜、结晶半导体膜和氧化物半导体膜等。
[0260]
另外,本实施方式所示的结构可以与其他实施方式所示的结构适当地组合而实施。
[0261]
实施方式2
[0262]
在本实施方式中,对包含嘧啶骨架的有机化合物进行说明。
[0263]
本发明的一个实施方式是由通式(g4)表示的有机化合物。
[0264][0265]
在通式(g4)中,ar
11
、ar
12
、r
11
至r
13
及r
21
至r
23
分别独立表示氢、碳数为1至4的烷基、取代或未取代的苯基和取代或未取代的联苯基中的任一个。另外,ar
13
及ar
14
分别独立表示
氢、碳数为1至4的烷基、取代或未取代的苯基、取代或未取代的联苯基、取代或未取代的二苯并噻吩-4-基和取代或未取代的二苯并呋喃-4-基中的任一个。另外,α1、α2、α8及α9分别独立表示取代或未取代的亚苯基。另外,h、i、x及y分别独立表示0或1。另外,e1及e2分别独立表示硫或氧。
[0266]
另外,本发明的一个实施方式是由通式(g5)表示的有机化合物。
[0267][0268]
在通式(g5)中,ar
11
、ar
12
、r
11
至r
13
及r
21
至r
23
分别独立表示氢、碳数为1至4的烷基、取代或未取代的苯基和取代或未取代的联苯基中的任一个。另外,ar
13
及ar
14
分别独立表示氢、碳数为1至4的烷基、取代或未取代的苯基、取代或未取代的联苯基、取代或未取代的二苯并噻吩-4-基和取代或未取代的二苯并呋喃-4-基中的任一个。另外,e1及e2分别独立表示硫或氧。
[0269]
另外,本发明的一个实施方式是由通式(g6)表示的有机化合物。
[0270][0271]
在通式(g6)中,r
11
至r
13
及r
21
至r
23
分别独立表示氢、碳数为1至4的烷基、取代或未取代的苯基和取代或未取代的联苯基中的任一个。另外,e1及e2分别独立表示硫或氧。
[0272]
另外,本发明的一个实施方式是由通式(g7)表示的有机化合物。
[0273][0274]
在通式(g7)中,r
11
至r
13
及r
21
至r
23
分别独立表示氢、碳数为1至4的烷基、取代或未取代的苯基和取代或未取代的联苯基中的任一个。另外,e1及e2分别独立表示硫或氧。
[0275]
另外,本发明的一个实施方式是由结构式(400)表示的有机化合物。
[0276][0277]
另外,作为通式(g4)及通式(g5)中的ar
13
及ar
14
的具体结构,例如可以举出由结构式(ar-6)至结构式(ar-11)表示的取代基。
[0278][0279]
另外,作为通式(g4)中的α1、α2、α8及α9的具体结构,例如可以举出由结构式(α-4)至结构式(α-6)表示的取代基。
[0280][0281]
当采用如结构式(α-4)那样的键合于对位的取代基的情况下,载流子传输性得到提高,所以是优选的。当采用如结构式(α-5)或结构式(α-6)那样的键合于间位或邻位的取代基的情况下,t1能级或s1能级得到提高,所以是优选的。
[0282]
另外,在ar
13
及ar
14
具有取代基的情况下,该取代基分别独立选自碳数为1至4的烷基、取代或未取代的苯基和取代或未取代的联苯基中的任一个。另外,在α1、α2、α8及α9具有取代基的情况下,该取代基分别独立选自碳数为1至4的烷基、取代或未取代的苯基、取代或未取代的联苯基、取代或未取代的二苯并噻吩-4-基和取代或未取代的二苯并呋喃-4-基中的任一个。另外,在通式(g4)至(g6)中,亚苯基键合到嘧啶环的2位、4位、5位和6位中的任一个或任两个。另外,在通式(g4)至(g7)中,作为取代基碳数为1至4的烷基、取代或未取代的苯基或取代或未取代的联苯基也可以键合到嘧啶环的2位、4位、5位和6位中的任一个或多
个。当具有这些取代基时,能够获得更立体的结构及稳定的膜性质,所以是优选的。另外,当具有烷基时,在溶剂中的溶解性得到提高,易于合成及利用湿法的成膜,所以是优选的。然而,当考虑合成成本时,有时优选不具有这些取代基。
[0283]
当上述通式(g4)及(g5)中的ar
11
及ar
12
为氢时,易于合成,所以是优选的。另外,当选择氢时,载流子(电子)注入性得到提高,能够期待低电压化,所以是优选的。另外,当ar
11
及ar
12
分别独立为碳数为1至4的烷基、取代或未取代的苯基或取代或未取代的联苯基时,非晶性得到提高,膜性质稳定,所以是优选的。
[0284]
作为由通式(g4)表示的有机化合物的具体例子,可以举出由结构式(400)至结构式(415)及结构式(430)至结构式(440)表示的有机化合物。注意,本发明不局限于这些化合物。
[0285]
[0286]
[0287]
[0288]
[0289][0290]
作为本发明的一个方式的有机化合物的合成方法可以应用各种反应。例如,通过进行下面描述的合成反应能够合成由通式(g4)表示的本发明的一个方式的有机化合物。另外,本发明的一个方式的有机化合物不局限于以下合成方法。
[0291]
《由通式(g4)表示的有机化合物的合成方法》
[0292]
首先,以下示出合成方案(b-1)。
[0293][0294]
如合成方案(b-1)所示,通过将二卤化嘧啶化合物(b1)与芳基硼化合物(b2)偶联,能够合成卤化嘧啶化合物(b3)。
[0295]
在合成方案(b-1)中,r
11
至r
13
分别独立表示氢、碳数为1至4的烷基、取代或未取代的苯基和取代或未取代的联苯基中的任一个。另外,ar
13
表示氢、碳数为1至4的烷基、取代或未取代的苯基、取代或未取代的联苯基、取代或未取代的二苯并噻吩-4-基和取代或未取代的二苯并呋喃-4-基中的任一个。另外,α1及α8分别独立表示取代或未取代的亚苯基。另外,h及x分别独立表示0或1。另外,e1表示硫或氧。另外,x3及x4分别独立表示氢、氯、溴或碘。注意,当ar
13
表示氢时,x4表示氢。另外,从高反应性的观点来看,x3及x4优选表示溴,更优选表示碘。另外,b3表示硼酸或二烷氧基硼。
[0296]
另外,多种反应条件可以用于合成方案(b-1)中的偶联反应。作为其中一个例子,可以采用在碱的存在下使用金属催化剂的合成方法。
[0297]
下面示出在合成方案(b-1)中采用铃木-宫浦反应的例子。可以使用钯催化剂作为金属催化剂,钯配合物与其配体的混合物可以用作钯催化剂。此外,作为钯配合物的例子,可以举出乙酸钯(ii)、四(三苯基膦)钯(0)和双(三苯基膦)二氯化钯(ii)等。作为配体的例子,可以举出三(邻甲苯基)膦、三苯基膦和三环己基膦等。此外,作为可以用作碱的物质的例子,可以举出有机碱如叔丁醇钠以及无机碱如碳酸钠或碳酸钾等。另外,所述反应优选在溶液中进行。可以使用的溶剂的例子如下所示:乙腈和水的混合溶剂;甲苯或二甲苯等稀释剂类和水的混合溶剂;甲苯或二甲苯、乙醇等醇和水的三种混合溶剂;1,3-二甲基-3,4,5,6-四氢-2(1h)-嘧啶酮(dmpu)和水的混合溶剂;乙二醇二甲基醚等醚类和水的混合溶剂等。但是,可以使用的催化剂、碱、溶剂不局限于这些。
[0298]
此外,在合成方案(b-1)中,可以使用芳基铝化合物、芳基锆化合物、芳基锌化合物或芳基锡化合物等代替芳基硼化合物(b2)。另外,反应优选在氮气、氩气等惰性气氛下进行。另外,也可以利用电磁波进行加热。
[0299]
接着,如合成方案(b-2)所示,通过将卤化嘧啶化合物(b3)与芳基硼化合物(b4)偶联可以合成本实施方式所示的由通式(g4)表示的有机化合物。
[0300][0301]
在合成方案(b-2)中,ar
11
、ar
12
、r
11
至r
13
及r
21
至r
23
分别独立表示氢、碳数为1至4的烷基、取代或未取代的苯基和取代或未取代的联苯基中的任一个。另外,ar
13
及ar
14
分别独立表示氢、碳数为1至4的烷基、取代或未取代的苯基、取代或未取代的联苯基、取代或未取代的二苯并噻吩-4-基和取代或未取代的二苯并呋喃-4-基中的任一个。另外,α1、α2、α8及α9分别独立表示取代或未取代的亚苯基。另外,h、i、x及y分别独立表示0或1。另外,e1及e2分别独立表示硫或氧。另外,x3表示氢、氯、溴或碘。注意,当ar
13
表示氢时,x3表示氢。另外,从高反应性的观点来看,x3优选表示溴,更优选表示碘。另外,b4表示硼酸或二烷氧基硼。
[0302]
另外,多种反应条件可以用于合成方案(b-2)中的偶联反应。作为其中一个例子,可以采用在碱的存在下使用金属催化剂的合成方法。
[0303]
在合成方案(b-2)中可以采用铃木-宫浦反应。详细内容可以参照上述合成方案(b-1),所以在此省略。
[0304]
另外,当化合物(b2)与化合物(b4)的芳基部分同样时,可以同时进行上述合成方案(b-1)和(b-2)的反应(换言之,能够一同将化合物(b2)及化合物(b4)加入有机化合物(b1)中发生反应,易于合成,所以是优选的。
[0305]
通过上述步骤可以合成本实施方式的有机化合物。
[0306]
由于本实施方式的有机化合物具有高s1能级、高t1能级及宽homo能级与lumo能级之间能隙(eg),因此通过在发光元件中将本实施方式的有机化合物用于使发光层中的发光物质分散的主体材料,可以获得高电流效率。尤其是,本实施方式的有机化合物适合用作分散磷光化合物的主体材料。此外,由于本实施方式的有机化合物是电子传输性高的物质,因此可以将其适用于发光元件中的电子传输层的材料。通过使用本实施方式的有机化合物,能够实现驱动电压低且电流效率高的发光元件。此外,通过使用该发光元件,能够获得耗电量低的发光装置、电子设备及照明装置。
[0307]
实施方式3
[0308]
在本实施方式中,参照图2a和图2b说明一种发光元件,其中将实施方式2所示的有机化合物用于发光层。
[0309]
在本实施方式中,参照图2a和图2b说明一种发光元件,该发光元件包含实施方式2所示的有机化合物的一个例子的由结构式(400)表示的4,6-双[3-(二苯并噻吩-4-基)苯基]嘧啶(简称:4,6mdbtp2pm-ii)。
[0310]
在本实施方式的发光元件中,至少具有发光层的el层插入在一对电极之间。el层除了发光层以外还可具有多个层。所述多个层以具有高载流子注入性质和具有高载流子传输性质的物质形成的层的组合形式堆叠,这样发光区远离电极形成,即载流子在远离电极的部分重新结合。在本说明书中,将包含具有高载流子注入性质或具有高载流子传输性质的物质的层称作功能层,该功能层具有载流子的注入或传输等的功能。作为功能层,可以使用空穴注入层、空穴传输层、电子注入层、电子传输层等。
[0311]
在图2a所示的本实施方式的发光元件中,el层1020具有发光层1130,该el层1020在一对电极即第一电极1010和第二电极1030之间。el层1020具有空穴注入层1110、空穴传输层1120、发光层1130、电子传输层1140和电子注入层1150。另外,图2a所示的发光元件具有:形成在衬底1000上的第一电极1010;在第一电极1010上依次形成的空穴注入层1110、空穴传输层1120、发光层1130、电子传输层1140和电子注入层1150;形成在电子注入层1150上的第二电极1030。注意,在本实施方式所述的发光元件中,第一电极1010用作阳极,第二电极1030用作阴极。
[0312]
衬底1000用作发光元件的支承。例如,可以使用玻璃、石英、塑料等作为衬底1000。或者,可以使用柔性衬底。柔性衬底是可以弯曲的衬底,例如由聚碳酸酯、聚芳酯或者聚醚砜制成的塑料衬底。或者,可以使用薄膜(由聚丙烯、聚酯、聚氟乙烯、聚氯乙烯等构成)、通过蒸镀形成的无机薄膜等。注意,可以使用其它材料,只要其在发光元件的制造过程中起支承物的作用。
[0313]
作为第一电极1010,优选使用具有高功函(具体地,4.0ev以上)的金属、合金或导电化合物或它们的混合物等。具体例子包括:氧化铟锡(ito:indium tin oxide)、含硅或氧化硅的氧化铟锡、氧化铟锌、含氧化钨和氧化锌的氧化铟(iwzo)等。这些导电金属氧化物的薄膜通常采用溅射法形成,但是也可以采用溶胶-凝胶法等形成。例如,氧化铟锌可采用溅射法使用在氧化铟中加入1wt%至20wt%氧化锌得到的靶材形成。另外,含有氧化钨及氧化锌的氧化铟(iwzo)可以采用溅射法使用在氧化铟中加入0.5wt%至5wt%氧化钨和0.1wt%至1wt%氧化锌得到的靶材形成。另外,还可以使用金、铂、镍、钨、铬、钼、铁、钴、铜、钯、金属氮化物材料(如氮化钛)等。
[0314]
注意,在el层1020中,当使用复合材料形成与第一电极1010接触的层时,所述复合材料使用有机化合物和下面描述的电子受体(acceptor)形成,可以使用各种金属、合金、导电化合物、它们的混合物等中的任何材料形成该第一电极1010,而不考虑功函。例如,可以使用铝、银、含铝合金(如al-si)等。
[0315]
形成在第一电极1010之上的el层1020至少包含发光层1130,el层1020的一部分包含为本发明的一个方式的有机化合物。el层1020的一部分可以使用已知物质,可以使用低分子化合物或高分子化合物。注意,另外,作为形成el层1020的物质,既可以采用只由有机化合物构成的物质,又可以采用其一部分含有无机化合物的物质。
[0316]
另外,如图2a所示,el层1020可通过适当叠加空穴注入层1110、空穴传输层1120、电子传输层1140、电子注入层1150等的组合以及发光层1130的方式形成。
[0317]
空穴注入层1110是包含具有高空穴注入性质的物质的层。具有高空穴注入性质的物质的例子包括:金属氧化物,如氧化钼、氧化钛、氧化钒、氧化铼、氧化钌、氧化铬、氧化锆、氧化铪、氧化钽、氧化银、氧化钨和氧化锰。或者可以使用酞菁基化合物如酞菁(简称:h2pc)或酞菁铜(ii)(简称:cupc)。
[0318]
或者,可以使用以下低分子量有机化合物:4,4
′
,4
″‑
三(n,n-二苯基氨基)三苯基胺(简称:tdata)、4,4
′
,4
″‑
三[n-(3-甲基苯基)-n-苯基氨基]三苯基胺(简称:mtdata)、4,4
′‑
二[n-(4-二苯基氨基苯基)-n-苯基氨基]联苯(简称:dpab)、4,4
′‑
二(n-{4-[n
′‑
(3-甲基苯基)-n
′‑
苯基氨基]苯基}-n-苯基氨基)联苯(简称:dntpd)、1,3,5-三[n-(4-二苯基氨基苯基)-n-苯基氨基]苯(简称:dpa3b)、3-[n-(9-苯基咔唑-3-基)-n-苯基氨基]-9-苯基咔唑(简称:pczpca1)、3,6-双[n-(9-苯基咔唑-3-基)-n-苯基氨基]-9-苯基咔唑(简称:pczpca2)和3-[n-(1-萘基)-n-(9-苯基咔唑-3-基)氨基]-9-苯基咔唑(简称:pczpcn1)等。
[0319]
另外,可以使用高分子化合物(例如,低聚物、枝状聚合物或聚合物)。高分子化合物的例子包括:聚(n-乙烯基咔唑)(简称:pvk)、聚(4-乙烯基三苯基胺)(简称:pvtpa)、聚[n-(4-{n
′‑
[4-(4-二苯基氨基)苯基]苯基-n
′‑
苯基氨基}苯基)甲基丙烯酰胺](简称:ptpdma)或聚[n,n
′‑
双(4-丁基苯基)-n,n
′‑
双(苯基)联苯胺](简称:poly-tpd)等。或者,还可以使用添加有酸的高分子化合物,例如,聚(3,4-亚乙基二氧噻吩)/聚(苯乙烯磺酸)(pedot/pss)或聚苯胺/聚(苯乙烯磺酸)(pani/pss)。
[0320]
可以将有机化合物与电子受体(acceptor)混合形成的复合材料用于空穴注入层1110。这种复合材料因为能通过电子受体在有机化合物中产生空穴而具有优异的空穴注入性质和空穴传输性质。在这种情况下,有机化合物优选是在传输产生的空穴方面性能优异的材料(具有高空穴传输性质的物质)。
[0321]
作为用于复合材料的有机化合物,可以使用各种化合物,例如芳族胺化合物、咔唑化合物、芳烃和高分子化合物(例如,低聚物、枝状聚合物或聚合物)。用于复合材料的有机化合物优选是具有高空穴传输性质的有机化合物。具体地,优选使用空穴迁移率10-6
cm2/vs以上的物质。注意,可以使用上述物质外的其他物质,只要这些物质是空穴传输性质大于电子传输性质的物质。可以用于复合材料的有机化合物的具体例子如下。
[0322]
可用于该复合材料的有机化合物的例子包括:芳族胺化合物如tdata、mtdata、dpab、dntpd、dpa3b、pczpca1、pczpca2、pczpcn1、4,4
′‑
双[n-(1-萘基)-n-苯基氨基]联苯(简称:npb或α-npd)、n,n
′‑
双(3-甲基苯基)-n,n
′‑
二苯基-[1,1
′‑
联苯]-4,4
′‑
二胺(简称:tpd)和4-苯基-4
′‑
(9-苯基芴-9-基)三苯基胺(简称:bpaflp);咔唑化合物如4,4
′‑
二(n-咔唑基)联苯(简称:cbp)、1,3,5-三[4-(n-咔唑基)苯基]苯(简称:tcpb)、9-[4-(10-苯基-9-蒽基)苯基]-9h-咔唑(简称:czpa)、9-苯基-3-[4-(10-苯基-9-蒽基)苯基]-9h-咔唑(简称:pczpa)和1,4-双[4-(n-咔唑基)苯基]-2,3,5,6-四苯基苯。
[0323]
或者,可以使用以下任意一种芳烃化合物:2-叔丁基-9,10-双(2-萘基)蒽(简称:t-budna)、2-叔丁基-9,10-二(1-萘基)蒽、9,10-二(3,5-二苯基苯基)蒽(简称:dppa)、2-叔丁基-9,10-双(4-苯基苯基)蒽(简称:t-budba)、9,10-二(2-萘基)蒽(简称:dna)、9,10-二苯基蒽(简称:dpanth)、2-叔丁基蒽(简称:t-buanth)、9,10-双(4-甲基-1-萘基)蒽(简称:
二苯基-9h-咔唑-3-胺(简称:2pcabpha)、n-(9,10-二苯基-2-蒽基)-n,n
′
,n
′‑
三苯基-1,4-亚苯基二胺(简称:2dpapa)、n-[9,10-双(1,1
′‑
联苯-2-基)-2-蒽基]-n,n
′
,n
′‑
三苯基-1,4-亚苯基二胺(简称:2dpabpha)、n-[9,10-双(1,1
′‑
联苯-2-基)]-n-[4-(9h-咔唑-9-基)苯基]-n-苯基蒽-2-胺(简称:2ygabpha)、n,n,9-三苯基蒽-9-胺(简称:dphapha)等。发射黄光的材料的例子包括:红荧烯、5,12-双(1,1
′‑
联苯-4-基)-6,11-二苯基并四苯(简称:bpt)等。另外,发射红光的材料的例子包括:n,n,n
′
,n
′‑
四(4-甲基苯基)并四苯-5,11-二胺(简称:p-mphtd)、7,14-二苯基-n,n,n
′
,n
′‑
四(4-甲基苯基)苊并(acenaphtho)[1,2-a]荧蒽-3,10-二胺(简称:p-mphafd)等。
[0334]
此外,在可以用于发光层1130的磷光化合物中,例如作为蓝色发光材料可以举出双[2-(4
′
,6
′‑
二氟苯基)吡啶合-n,c2′
]铱(ⅲ)四(1-吡唑基)硼酸盐(简称:fir6)、双[2-(4
′
,6
′‑
二氟苯基)吡啶合-n,c2′
]铱(ⅲ)吡啶甲酸酯(简称:firpic)、双{2-[3
′
,5
′‑
双(三氟甲基)苯基]吡啶合-n,c2′
}铱(ⅲ)吡啶甲酸酯(简称:ir(cf3ppy)2(pic))、双[2-(4
′
,6
′‑
二氟苯基)吡啶合-n,c2′
]铱(ⅲ)乙酰丙酮(简称:fir(acac))等。另外,发射绿光的材料的例子包括:三(2-苯基吡啶合-n,c2′
)铱(iii)(简称:ir(ppy)3)、乙酰丙酮双(2-苯基吡啶-n,c2′
)铱(iii)(简称:ir(ppy)2(acac))、乙酰丙酮双(1,2-二苯基-1h-苯并咪唑合)铱(iii)(简称:ir(pbi)2(acac))、乙酰丙酮双(苯并[h]喹啉合)铱(iii)(简称:ir(bzq)2(acac))、三(苯并[h]喹啉合)铱(iii)(简称:ir(bzq)3)等。发射黄光的材料的例子包括:乙酰丙酮双(2,4-二苯基-1,3-唑合-n,c2′
)铱(iii)(简称:ir(dpo)2(acac))、乙酰丙酮双[2-(4
′‑
(五氟苯基苯基)吡啶合]铱(iii)(简称:ir(p-pf-ph)2(acac))、乙酰丙酮双(2-苯基苯并噻唑合-n,c2′
)铱(iii)(简称:ir(bt)2(acac))、(乙酰丙酮)双[2,3-双(4-氟苯基)-5-甲基吡嗪合]铱(ⅲ)(简称:ir(fdppr-me)2(acac))、(乙酰丙酮)双{2-(4-甲氧基苯基)-3,5-二甲苯吡嗪合}铱(ⅲ)(简称:ir(dmmoppr)2(acac))等。发射橙色光的材料的例子包括:三(2-苯基喹啉合-n,c2′
)铱(iii)(简称:ir(pq)3)、乙酰丙酮双(2-苯基喹啉合-n,c2′
)铱(iii)(简称:ir(pq)2(acac))、(乙酰丙酮)双(3,5-二甲基-2-苯基吡嗪合)铱(ⅲ)(简称:ir(mppr-me)2(acac))、(乙酰丙酮)双(5-异丙基-3-甲基-2-苯基吡嗪合)铱(ⅲ)(简称:ir(mppr-ipr)2(acac))等。发射红光的材料的例子包括:有机金属配合物,例如,乙酰丙酮双[2-(2
′‑
苯并[4,5-α]噻吩基)吡啶-n,c3′
)铱(iii)(简称:ir(btp)2(acac))、乙酰丙酮双(1-苯基异喹啉合-n,c2′
)铱(iii)(简称:ir(piq)2(acac)、(乙酰丙酮)双2,3-二(4-氟苯基)喹喔啉合]铱(iii)(简称:ir(fdpq)2(acac))、(乙酰丙酮)双[2,3,5-三苯基吡嗪合]铱(iii)(简称:ir(tppr)2(acac))、双(2,3,5-三苯基吡嗪合)(二新戊酰基甲烷合)铱(iii)(简称:ir(tppr)2(dpm))和2,3,7,8,12,13,17,18-八乙基-21h,23h-卟啉铂(ii)(简称:ptoep)。另外,因为通过例如以下的稀土金属配合物可得到由稀土金属离子发射光(在不同多重性之间的电子跃迁):三(乙酰丙酮)(单菲咯啉)铽(iii)(简称:tb(acac)3(phen))、三(1,3-二苯基-1,3-丙二酸(propanedionato))(单菲咯啉)铕(iii)(简称:eu(dbm)3(phen))和三[1-(2-噻吩甲酰基)-3,3,3-三氟丙酮合](单菲咯啉)铕(iii)(简称:eu(tta)3(phen)),这类稀土金属配合物可以用作磷光化合物。
[0335]
作为发光物质,还可以使用高分子化合物。具体地,发射蓝光的材料的例子包括:聚(9,9-二辛基芴-2,7-二基)(简称:pfo)、[(9,9-二辛基芴-2,7-二基)-(2,5-二甲氧基苯-1,4-二基)]共聚物(简称:pf-dmop)、{(9,9-二辛基芴-2,7-二基)-[n,n
′‑
二-(对丁基苯
基)-1,4-二氨基苯]}(简称:tab-pfh)等。另外,发射绿光的材料的例子包括:聚(对亚苯基亚乙烯基)(简称:ppv)、[(9,9-二己基芴-2,7-二基)-(苯并[2,1,3]噻二唑-4,7-二基)交替共聚物](简称:pfbt)、[(9,9-二辛基-2,7-二亚乙烯基亚芴基(fluorenylene))-(2-甲氧基-5-(2-乙基己氧基)-1,4-亚苯基)交替共聚物等。另外,发射橙光至红光的材料的例子包括:聚[2-甲氧基-5-(2
′‑
乙基己氧基)-1,4-亚苯基亚乙烯基](简称:meh-ppv)、聚(3-丁基噻吩-2,5-二基)(简称:r4-pat)、{[9,9-二己基-2,7-二(1-氰基亚乙烯基)亚芴基]-[2,5-双(n,n
′‑
二苯基氨基)-1,4-亚苯基]}交替共聚物、{[2-甲氧基-5-(2-乙基己氧基)-1,4-双(1-氰基亚乙烯基亚苯基)]-[2,5-双(n,n
′‑
二苯基氨基)-1,4-亚苯基]}交替共聚物(简称:cn-ppv-dpd)等。
[0336]
另外,实施方式2所示的有机化合物及实施方式1所示的第二化合物也具有荧光性,所以可用作发光材料。
[0337]
电子传输层1140是包含具有高电子传输性质的物质的层。作为具有高电子传输性质的物质,可以使用例如以下物质:具有喹啉骨架或苯并喹啉骨架的金属配合物,例如三(8-羟基喹啉)铝(简称:alq)、三(4-甲基-8-羟基喹啉)铝(简称:almq3)、双(10-羟基苯并[h]-喹啉)铍(简称:bebq2)或双(2-甲基-8-羟基喹啉)(4-苯基苯酚)铝(简称:balq)。或者可以使用包含唑基配体或噻唑基配体的金属配合物等,例如双[2-(2-羟基苯基)苯并 唑合]锌(简称:zn(box)2)或二[2-(2-羟基苯基)苯并噻唑合]锌(简称:zn(btz)2)。还可以使用金属配合物外的其他化合物:2-(4-联苯基)-5-(4-叔丁基苯基)-1,3,4
‑ꢀ
二唑(简称:pbd)、1,3-双[5-(对-叔丁基苯基)-1,3,4
‑ꢀ
二唑-2-基]苯(简称:oxd-7)、3-(4-联苯基)-4-苯基-5-(4-叔丁基苯基)-1,2,4-三唑(简称:taz)、红菲绕啉(简称:bphen)、浴铜灵(简称:bcp)等。在此所述的物质主要是电子迁移率10-6
cm2/vs以上的物质。另外,所述电子传输层不限于单层,可以是包含所述物质的两个或多个层的叠层。
[0338]
另外,实施方式2所示的有机化合物及实施方式1所示的第二化合物也具有嘧啶骨架,所以适用于电子传输层1140。
[0339]
电子注入层1150是包含具有高电子注入性质的物质的层。对电子注入层1150,可以使用碱金属、碱土金属或者它们的化合物,例如锂、铯、钙、氟化锂、氟化铯、氟化钙或者氧化锂。或者,可以使用稀土金属化合物如氟化铒。或者,还可以使用上述形成电子传输层1140的物质。
[0340]
或者,可以将有机化合物与电子给体(donor)混合形成的复合材料用于电子注入层1150。这种复合材料因为能通过电子供体在有机化合物中产生电子而具有优异的电子注入性质和电子传输性质。在这种情况下,有机化合物优选是在传输产生的电子方面性能优异的材料。具体地,例如,可以使用上面所述的形成电子传输层1140的物质(如,金属配合物和杂芳族化合物)。作为电子给体,可使用对有机化合物显示电子给体性质的物质。具体地,优选使用碱金属、碱土金属和稀土金属,例如锂、铯、镁、钙、铒、镱等。另外,优选使用碱金属氧化物或碱土金属氧化物,例如氧化锂、氧化钙、氧化钡等。或者可以使用路易斯碱如氧化镁。或者可以使用四硫富瓦烯(简称:ttf)等有机化合物。
[0341]
注意,上述各空穴注入层1110、空穴传输层1120、发光层1130、电子传输层1140和电子注入层1150可采用如蒸镀法(包括真空蒸镀法)、喷墨法或涂覆法形成。
[0342]
当第二电极1030用作阴极时,该电极优选使用具有低功函(优选,功函3.8ev以下)
的金属、合金、导电化合物以及它们的混合物而形成。具体地,可以使用:铝或银;属于周期表第1族或第2族的元素,即锂或铯等碱金属、钙或锶等碱土金属;镁;上述金属的合金(如mg-ag或al-li);铕或镱等稀土金属;上述金属的合金等。
[0343]
注意,在el层1020中,与第二电极1030接触形成的层是由复合材料形成时,可以使用各种导电材料如铝、银、ito、含硅或氧化硅的氧化铟锡,而不考虑功函,所述复合材料是上面所述的有机化合物与电子给体混合形成。
[0344]
注意,第二电极1030可以采用真空蒸镀法或溅射法形成。或者,在使用银糊等的情况下,可以采用涂覆法、喷墨法等。
[0345]
在上述发光元件中,因为在第一电极1010和第二电极1030之间产生的电位差而产生电流,因为在el层1020中空穴和电子重新结合而发光。然后发射的光通过第一电极1010和第二电极1030中的一方或者双方被取出到外部。因此,第一电极1010和第二电极1030中的一方或双方是具有透射可见光性质的电极。
[0346]
另外,在第一电极1010和第二电极1030之间的层结构不限于上述结构。可以采用不同于上述的结构,只要在远离第一电极1010和第二电极1030的部分中具有空穴和电子重新结合的发光区,以防止发光区接近金属而产生的猝灭。
[0347]
也就是说,对层的叠层结构没有特别的限制。使用高电子传输性质的物质、高空穴传输性质的物质、高电子注入性质的物质和具有高空穴注入性质的物质、双极物质(具有高电子传输性质和高空穴传输性质的物质)、空穴阻挡物质等可以与发光层自由组合,所述发光层包含本发明的一个方式的4,6mdbtp2pm-ii作为主体材料。
[0348]
注意,因为4,6mdbtp2pm-ii是具有高电子传输性质的物质,所以作为电子传输层1140可以使用4,6mdbtp2pm-ii。即,本发明的一个方式的有机化合物可以用于电子传输层。
[0349]
另外,通过将本发明的一个方式的有机化合物应用于发光层1130(尤其是发光层的主体材料)和电子传输层1140的双方,可以实现极低的驱动电压。
[0350]
另外,在图2b所示的发光元件中,el层1020设置在衬底1000上的一对电极即第一电极1010和第二电极1030之间。el层1020具有空穴注入层1110、空穴传输层1120、发光层1130、电子传输层1140和电子注入层1150。图2b所示的发光元件具有:衬底1000上的用作阴极的第二电极1030;在第二电极1030上依次层叠的电子注入层1150,电子传输层1140,发光层1130,空穴传输层1120和空穴注入层1110;以及空穴注入层1110上的用作阳极的第一电极1010。
[0351]
下面具体描述形成发光元件的方法。
[0352]
本实施方式的发光元件具有将el层1020插在一对电极之间的结构。所述el层1020至少具有发光层1130,所述发光层1130使用4,6mdbtp2pm-ii作为主体材料而形成。另外,el层1020除了具有发光层1130外还可以具有功能层(如,空穴注入层1110、空穴传输层1120、电子传输层1140或电子注入层1150)。各电极(第一电极1010或第二电极1030)、发光层1130和各功能层可以采用湿法或者干法形成,所述湿法例如有滴液排放法(喷墨法)、旋涂法或印刷法,干法例如有真空蒸镀法、cvd法或溅射法。湿法能够采用简单的装置和方法在常压下进行,因此具有简化工艺和提高生产率的效应。与湿法不同,干法不需要将材料溶解,能够使用在溶液中低溶解度的材料,因此扩大材料的选择范围。
[0353]
发光元件中包含的所有薄膜都可以采用湿法形成。在这种情况下,发光元件可以
只用湿法要求的设备制造。或者,形成层叠的层至形成发光层1130可以采用湿法进行,而堆叠在发光层1130上的功能层、第一电极1010等可采用干法形成。或者,在形成发光层1130之前采用干法形成第二电极1030和功能层,而发光层1130、堆叠在其上的功能层以及第一电极1010可以采用湿法形成。不必说,本实施方式不限于此,可以根据使用的材料、必需的膜厚度和界面状态,适当选择湿法或干法形成发光元件。
[0354]
在本实施方式中,可以在玻璃、塑料等构成的衬底上制造发光元件。通过在一个衬底上形成多个所述发光元件,可以制造被动矩阵发光装置。或者,在玻璃、塑料等构成的衬底上形成例如薄膜晶体管(tft),在与tft电连接的电极上可以制造发光元件。因此,制造主动矩阵发光装置,该装置中tft控制发光元件的驱动。注意,对tft的结构没有特别的限制。可以使用交错tft(staggered tft)或反转交错tft。另外,对用于tft的半导体的结晶性没有特别的限制;可以使用非晶半导体或者晶体半导体。另外,在tft衬底上形成的驱动电路可以由n型和p型tft中的一方或双方形成。
[0355]
如上所述,可以使用实施方式2中所述的4,6mdbtp2pm-ii来制造发光元件。通过将本发明的一个方式的有机化合物用于发光元件,可以得到驱动电压低且电流效率高的发光元件。
[0356]
另外,使用按照上述方法得到的本发明的一个方式的发光元件的发光装置(图像显示装置)可实现低耗电量。
[0357]
注意,通过使用在本实施方式中所述的发光元件,可以制造被动矩阵发光装置或通过薄膜晶体管(tft)控制发光元件的驱动的主动矩阵发光装置。
[0358]
本实施方式可以与其他实施方式适当组合实施。
[0359]
实施方式4
[0360]
在本实施方式中,作为本发明的一个方式参照图3说明一种发光元件,其中将第一化合物的磷光铱金属配合物、第二化合物的包含嘧啶骨架的有机化合物以及其他两种以上的有机化合物用于发光层。
[0361]
本实施方式所示的发光元件具有如图3所示那样的在一对电极(第一电极201与第二电极202)之间具有el层203的结构。另外,el层203至少具有发光层204,除此之外,el层203还可以具有空穴注入层、空穴传输层、电子传输层、电子注入层、电荷产生层等。此外,作为空穴注入层、空穴传输层、电子传输层、电子注入层及电荷产生层可以使用实施方式1所示的物质。另外,在本实施方式中,第一电极201用作阳极,第二电极202用作阴极。
[0362]
本实施方式所示的发光层204包含使用实施方式1所示的第一化合物的磷光铱金属配合物的磷光化合物205、第一有机化合物206及第二有机化合物207。另外,磷光化合物205是发光层204中的客体材料。此外,第一有机化合物206和第二有机化合物207中的至少一方含有第二化合物的包含嘧啶骨架的有机化合物,其中发光层204中的含有率多的一方用作发光层204的主体材料。
[0363]
在发光层204中,通过采用将上述客体材料分散到主体材料的结构,可以控制发光层的结晶化。另外,可以抑制由于客体材料的高浓度导致的浓度猝灭并提高发光元件的发光效率。
[0364]
另外,第一有机化合物206和第二有机化合物207的每一个的三重激发态能的能级(t1能级)优选高于磷光化合物205的t1能级。这是因为如下缘故:如果第一有机化合物206
(或第二有机化合物207)的t1能级低于磷光化合物205的t1能级,则第一有机化合物206(或第二有机化合物207)使有助于发光的磷光化合物205的三重激发态能猝灭(quench),而导致发光效率的降低。
[0365]
在此,为了提高从主体材料到客体材料的能量转移效率,优选的是,在考虑到作为分子之间的转移机理周知的福斯特机理(偶极-偶极相互作用)及德克斯特(dexter)机理(电子交换相互作用)的情况下,主体材料的发射光谱(从单重激发态的能量转移中的荧光光谱,从三重激发态的能量转移中的磷光光谱)与客体材料的吸收光谱(更详细地说,最长波长(低能量)一侧的吸收带中的光谱)的重叠的部分大。但是,在采用一般的磷光主体材料的情况下,难以使主体材料的荧光光谱与客体材料的最长波长(低能量)一侧的吸收带中的光谱重叠。这是因为如下缘故:因为主体材料的磷光光谱位于比荧光光谱长的波长(低能量)一侧,所以如果那样做,则主体材料的t1能级低于磷光化合物的t1能级,而导致上述猝灭的问题。另一方面,为了避免猝灭的问题,当将主体材料的t1能级设定为高于磷光化合物的t1能级时,主体材料的荧光光谱漂移到短波长(高能量)一侧,因此该荧光光谱不与客体材料的最长波长(低能量)一侧的吸收带中的光谱重叠。从而,通常,难以将主体材料的荧光光谱与客体材料的最长波长(低能量)一侧的吸收带中的吸收光谱重叠并最大限度地提高主体材料的从单重激发态的能量转移。
[0366]
于是,在本实施方式中,第一有机化合物206和第二有机化合物207的组合优选是形成激基复合物的组合。此时,当在发光层204中载流子(电子及空穴)重新结合时,第一有机化合物206和第二有机化合物207形成激基复合物(也称为“exciplex”)。由此,在发光层204中,第一有机化合物206的荧光光谱及第二有机化合物207的荧光光谱转换为位于更长波长一侧的激基复合物的发射光谱。并且,如果为了使激基复合物的发射光谱与客体材料的吸收光谱的重叠的部分变大而选择第一有机化合物和第二有机化合物,则可以最大限度地提高从单重激发态的能量转移。另外,关于三重激发态,也可以认为产生来自激基复合物的能量转移,而不产生来自主体材料的能量转移。
[0367]
作为磷光化合物205,使用第一化合物的磷光铱金属配合物。另外,作为第一有机化合物206和第二有机化合物207的组合,优选组合容易接受电子的化合物(电子俘获化合物)和容易接受空穴的化合物(空穴俘获化合物)。另外,第二化合物的包含嘧啶骨架的有机化合物可以用作容易接受电子的化合物。
[0368]
作为容易接受空穴的化合物,例如可以举出4-苯基-4
′‑
(9-苯基-9h-咔唑-3-基)三苯胺(简称:pcba1bp)、3-[n-(1-萘基)-n-(9-苯基咔唑-3-基)氨基]-9-苯基咔唑(简称:pczpcn1)、4,4
′
,4
″‑
三[n-(1-萘基)-n-苯基氨基]三苯胺(简称:1
′‑
tnata)、2,7-双[n-(4-二苯基氨基苯基)-n-苯基氨基]-螺环-9,9
′‑
联芴(简称:dpa2sf)、n,n
′‑
双(9-苯基咔唑-3-基)-n,n
′‑
二苯基-苯-1,3-二胺(简称:pca2b)、n-(9,9-二甲基-2-n,n
′‑
二苯基氨基-9h-芴-7-基)-n,n-二苯基胺(简称:dpnf)、n-苯基-n-(4-苯基苯基)-n-(9-苯基-9h-咔唑-3-基)胺(简称:pca1bp)、n,n
′
,n
″‑
三苯基-n,n
′
,n
″‑
三(9-苯基咔唑-3-基)-苯-1,3,5-三胺(简称:pca3b)、2-[n-(9-苯基咔唑-3-基)-n-苯基氨基]-螺环-9,9
′‑
联芴(简称:pcasf)、2-[n-(4-二苯基氨基苯基)-n-苯基氨基]-螺环-9,9
′‑
联芴(简称:dpasf)、n,n-二(联苯-4-基)-n-(9-苯基-9h-咔唑-3-基)胺(简称:pczbba1)、n,n
′‑
双[4-(咔唑-9-基)苯基]-n,n
′‑
二苯基-9,9-二甲基芴-2,7-二胺(简称:yga2f)、4,4
′‑
双[n-(3-甲基苯基)-n-苯基氨基]联
苯(简称:tpd)、4,4
′‑
双[n-(4-二苯基氨基苯基)-n-苯基氨基]联苯(简称:dpab)、n-(9,9-二甲基-9h-芴-2-基)-n-[9,9-二甲基-2-{n
′‑
苯基-n
′‑
(9,9-二甲基-9h-芴-2-基)}氨基-9h-芴-7-基]苯基胺(简称:dfladfl)、3-[n-(9-苯基咔唑-3-基)-n-苯基氨基]-9-苯基咔唑(简称:pczpca1)、3-[n-(4-二苯基氨基苯基)-n-苯基氨基]-9-苯基咔唑(简称:pczdpa1)、3,6-双[n-(4-二苯基氨基苯基)-n-苯基氨基]-9-苯基咔唑(简称:pczdpa2)、4,4
′‑
双(n-{4-[n
′‑
(3-甲基苯基)-n
′‑
苯基氨基]苯基}-n-苯基氨基)联苯(简称:dntpd)、3,6-双[n-(4-二苯基氨基苯基)-n-(1-萘基)氨基]-9-苯基咔唑(简称:pcztpn2)、3,6-双[n-(9-苯基咔唑-3-基)-n-苯基氨基]-9-苯基咔唑(简称:pczpca2)。
[0369]
上述第一有机化合物206和第二有机化合物207的组合是能够形成激基复合物的组合的一例,该激基复合物的发射光谱与磷光化合物205的吸收光谱重叠,与磷光化合物205的吸收光谱的峰值相比,该激基复合物的发射光谱的峰值位于长波长,即可。
[0370]
另外,在由容易接受电子的化合物和容易接受空穴的化合物构成第一有机化合物206和第二有机化合物207时,可以根据其混合比控制载流子平衡。具体而言,优选第一有机化合物和第二有机化合物的比例为1:9至9:1。
[0371]
因为本实施方式所示的发光元件能够由于利用激基复合物的发射光谱与磷光化合物的吸收光谱的重叠的能量转移而提高能量转移效率,所以可以实现外量子效率高的发光元件。
[0372]
另外,作为包括在本发明中的其他结构,也可以采用如下结构:磷光化合物205(客体材料)以外的两种有机化合物使用具有空穴俘获性的主体分子及具有电子俘获性的主体分子来形成发光层204,以便得到将空穴和电子导入到存在于两种主体分子中的客体分子而使客体分子成为激发态的现象(即,guest coupled with complementary hosts:gcch,客体与互补主体的耦合)。
[0373]
此时,作为具有空穴俘获性的主体分子及具有电子俘获性的主体分子,分别可以使用上述容易接受空穴的化合物及容易接受电子的化合物。
[0374]
另外,本实施方式所示的发光元件是发光元件的结构的一个例子,但是也可以将其他实施方式所示的其他结构的发光元件应用于本发明的一个方式的发光装置。此外,作为具有上述发光元件的发光装置,可以制造被动矩阵型发光装置和主动矩阵型发光装置。还可以制造在其他实施方式所说明的具有与上述发光元件不同的发光元件的微腔结构的发光装置。上述发光装置都包括在本发明中。
[0375]
另外,在采用主动矩阵型发光装置的情况下,对tft的结构没有特别的限制。例如,可以适当地使用交错型tft或反交错型tft。此外,形成在tft衬底上的驱动电路可以由n型tft和p型tft中的一方或双方形成。并且,对用于tft的半导体膜的结晶性也没有特别的限制。例如,可以使用非晶半导体膜、结晶半导体膜和氧化物半导体膜等。
[0376]
另外,本实施方式所示的结构可以与其他实施方式所示的结构适当地组合而实施。
[0377]
实施方式5
[0378]
在本实施方式中,作为本发明的一个方式,对隔着电荷产生层具有多个el层的结构的发光元件(以下,称为串联型发光元件)进行说明。
[0379]
本实施方式所示的发光元件是如图4a所示那样的在一对电极(第一电极301与第
二电极304)之间具有多个el层(第一el层302(1)和第二el层302(2))的串联型发光元件。
[0380]
在本实施方式中,第一电极301是用作阳极的电极,第二电极304是用作阴极的电极。另外,作为第一电极301及第二电极304,可以采用与实施方式1相同的结构。此外,虽然多个el层(第一el层302(1)和第二el层302(2))可以具有与实施方式1或实施方式3所示的结构相同的结构,但是也可以上述el层中的任一个具有与实施方式1或实施方式3所示的结构相同的结构。换言之,第一el层302(1)和第二el层302(2)既可以具有相同结构,又可以具有不同的结构,作为其结构,可以应用与实施方式1或实施方式3相同的结构。
[0381]
另外,在多个el层(第一el层302(1)和第二el层302(2))之间设置有电荷产生层305。电荷产生层305具有如下功能:当对第一电极301和第二电极304施加电压时,将电子注入到一方的el层中,且将空穴注入到另一方的el层中。在本实施方式中,当对第一电极301施加电压以使其电位高于第二电极304时,电荷产生层305将电子注入到第一el层302(1)中,并将空穴注入到第二el层302(2)中。
[0382]
另外,从光提取效率的观点来看,电荷产生层305优选具有透射可见光的性质(具体而言,电荷产生层305具有40%以上的可见光透射率)。另外,电荷产生层305即使在其电导率小于第一电极301或第二电极304的情况下也发挥作用。
[0383]
电荷产生层305既可以具有对空穴传输性高的有机化合物添加有电子受体(受体)的结构,又可以具有对电子传输性高的有机化合物添加有电子给体(供体)的结构。或者,也可以层叠有这两种结构。
[0384]
在采用对空穴传输性高的有机化合物添加有电子受体的结构的情况下,作为空穴传输性高的有机化合物,例如可以使用芳族胺化合物诸如npb、tpd、tdata、mtdata或4,4
′‑
双[n-(螺-9,9
′‑
联芴-2-基)-n-苯基氨基]联苯(简称:bspb)等。在此所述的物质主要是空穴迁移率为10-6
cm2/vs以上的物质。但是,只要是空穴传输性比电子传输性高的有机化合物,就可以使用上述物质之外的物质。
[0385]
另外,作为电子受体,可以举出7,7,8,8-四氰基-2,3,5,6-四氟喹啉并二甲烷(简称:f
4-tcnq)、氯醌等。另外,还可以举出过渡金属氧化物。另外,可以举出属于元素周期表中第4族至第8族的金属的氧化物。具体而言,优选使用氧化钒、氧化铌、氧化钽、氧化铬、氧化钼、氧化钨、氧化锰和氧化铼,这是因为它们具有高电子接受性。尤其,优选使用氧化钼,因为氧化钼在大气中稳定且其吸湿性低,所以容易进行处理。
[0386]
另一方面,在采用对电子传输性高的有机化合物添加有电子给体的结构的情况下,作为电子传输性高的有机化合物,例如可以使用具有喹啉骨架或苯并喹啉骨架的金属配合物等诸如alq、almq3、bebq2或balq等。除此之外,还可以使用具有 唑基配体或噻唑基配体的金属配合物等诸如zn(box)2或zn(btz)2等。再者,除了金属配合物之外,还可以使用pbd、oxd-7、taz、bphen、bcp等。在此所述的物质主要是电子迁移率为10-6
cm2/vs以上的物质。此外,也可以使用第二化合物的包含嘧啶骨架的有机化合物。另外,只要是电子传输性比空穴传输性高的有机化合物,就可以使用上述物质之外的物质。
[0387]
另外,作为电子给体,可以使用碱金属、碱土金属、稀土金属、属于元素周期表中第2族或第13族的金属及它们的氧化物和碳酸盐。具体而言,优选使用锂(li)、铯(cs)、镁(mg)、钙(ca)、镱(yb)、铟(in)、氧化锂、碳酸铯等。此外,也可以将如四硫萘并萘(tetrathianaphthacene)等的有机化合物用作电子给体。
[0388]
另外,通过使用上述材料形成电荷产生层305,可以抑制el层层叠时造成的驱动电压的增大。
[0389]
虽然在本实施方式中,对具有两个el层的发光元件进行说明,但是,如图4b所示那样,本发明的一个方式可以同样地应用于层叠n个(注意,n是3以上)el层的发光元件。如根据本实施方式的发光元件那样,当在一对电极之间具有多个el层时,通过将电荷产生层设置在el层与el层之间,可以在保持低电流密度的同时实现高亮度区域中的发光。因为可以保持低电流密度,所以可以实现长寿命元件。另外,当将该发光元件应用于照明时,因为可以减少由于电极材料的电阻导致的电压下降,所以可以实现大面积的均匀发光。此外,可以实现能够进行低电压驱动且耗电量低的发光装置。
[0390]
此外,通过使各el层发射互不相同颜色的光,可以使发光元件整体发射所需颜色的光。例如,在具有两个el层的发光元件中,使第一el层的发光颜色和第二el层的发光颜色处于补色关系,因此发光元件作为整体可以得到发射白色发光。注意,词语“补色关系”表示当颜色混合时得到非彩色的颜色关系。也就是说,可以通过混合从发射补色光的物质中得到的光来得到白色光。
[0391]
另外,具有三个el层的发光元件的情况也与此同样,例如,当第一el层的发射色是红色,第二el层的发射色是绿色,第三el层的发射色是蓝色时,发光元件作为整体可以得到白色发光。
[0392]
另外,本实施方式所示的结构可以与其他实施方式所示的结构适当地组合而实施。
[0393]
实施方式6
[0394]
在本实施方式中,参照图5对使用实施方式1及实施方式3至实施方式5所示的发光元件的发光装置进行说明。
[0395]
本实施方式所示的发光装置具有利用一对电极之间的光的共振效应的光学微谐振腔(micro optical resonator)(微腔)结构,如图5所示具有多个发光元件,该发光元件具有在一对电极(反射电极451与半透过半反射电极452)之间至少具有el层455的结构。另外,el层455至少具有用作发光区的第一发光层454b、第二发光层454g及第三发光层454r,除此之外,el层455还可以具有空穴注入层、空穴传输层、电子传输层、电子注入层、电荷产生层等。此外,第一发光层454b、第二发光层454g及第三发光层454r中的至少一层包含磷光铱金属配合物及包含嘧啶骨架的有机化合物。
[0396]
在本实施方式中,对如图5所示那样具有结构不同的发光元件(第一发光元件450r、第二发光元件450g和第三发光元件450b)的发光装置进行说明。
[0397]
第一发光元件450r具有在反射电极451上依次层叠有如下层的结构:第一透明导电层453a;el层455;以及半透射半反射电极452。另外,第二发光元件450g具有在反射电极451上依次层叠有第二透明导电层453b、el层455以及半透射半反射电极452的结构。另外,第三发光元件450b具有在反射电极451上依次层叠有el层455及半透射半反射电极452的结构。
[0398]
另外,上述发光元件(第一发光元件450r、第二发光元件450g及第三发光元件450b)都具有反射电极451、el层455以及半透射半反射电极452。
[0399]
另外,el层455包含第一发光层454b、第二发光层454g和第三发光层454r。此外,第
一发光层454b发射在420nm以上且480nm以下的波长区域中具有峰值的光(λb),第二发光层454g发射在500nm以上且550nm以下的波长区域中具有峰值的光(λg),而第三发光层454r发射在600nm以上且760nm以下的波长区域中具有峰值的光(λr)。由此,可以使任何发光元件(第一发光元件450r、第二发光元件450g及第三发光元件450b)都发射将来自第一发光层454b、第二发光层454g及第三发光层454r的发光重叠而成的光,即及于可见光区的发射光谱宽的光。注意,根据上述记载,波长的长度满足λb《λg《λr的关系。
[0400]
本实施方式所示的各发光元件分别具有在反射电极451与半透射半反射电极452之间夹有el层455的结构,并且从包括在el层455中的各发光层向全方向射出的光由具有光学微谐振腔(微腔)的功能的反射电极451和半透射半反射电极452共振。另外,反射电极451使用具有反射性的导电材料形成,可见光的对该膜的反射率为40%至100%,优选为70%至100%,并且该膜的电阻率为1
×
10-2
ωcm以下。另外,半透射半反射电极452使用具有反射性的导电材料和具有透光性的导电材料形成,可见光的对该膜的反射率为20%至80%,优选为40%至70%,并且该膜的电阻率为1
×
10-2
ωcm以下。
[0401]
另外,在本实施方式中,通过使分别设置在第一发光元件450r和第二发光元件450g中的透明导电层(第一透明导电层453a、第二透明导电层453b)的厚度彼此不同,来根据每个发光元件改变反射电极451与半透射半反射电极452之间的光程。换言之,在反射电极451与半透射半反射电极452之间,可以使从各发光元件的各发光层发射的发射光谱宽的光中的共振的波长的光变强并可以使其中的不共振的波长的光衰减,所以通过根据每个元件改变反射电极451与半透射半反射电极452之间的光程,可以取出不同波长的光。
[0402]
另外,光程(也称为光路长度)是指实际上的距离乘以折射率而求得的值,在本实施方式中,表示实际上的厚度乘以n(折射率)而求得的值。换言之,“光程=实际上的厚度
×
n”。
[0403]
另外,在第一发光元件450r中从反射电极451到半透射半反射电极452的光程为mλr/2(注意,m是1以上的自然数),在第二发光元件450g中从反射电极451到半透射半反射电极452的光程为mλg/2(注意,m是1以上的自然数),并且在第三发光元件450b中从反射电极451到半透射半反射电极452的光程为mλb/2(注意,m是1以上的自然数)。
[0404]
如上所述,从第一发光元件450r主要取出在包括于el层455中的第三发光层454r中发射的光(λr),从第二发光元件450g主要取出在包括于el层455中的第二发光层454g中发射的光(λg),并且从第三发光元件450b主要取出在包括于el层455中的第一发光层454b中发射的光(λb)。另外,从各发光元件取出的光分别从半透射半反射电极452一侧射出。
[0405]
另外,在上述结构中,严格而言,将从反射电极451到半透射半反射电极452的光程称为从反射电极451中的反射区域到半透射半反射电极452中的反射区域的距离。但是,难以严格地决定反射电极451或半透射半反射电极452中的反射区域的位置,所以通过假定反射电极451和半透射半反射电极452中的任意的位置为反射区域来可以充分地获得上述效果。
[0406]
接着,在第一发光元件450r中,因为来自第三发光层454r的发光中的由反射电极451反射而回来的光(第一反射光)与从第三发光层454r直接入射到半透射半反射电极452的光(第一入射光)发生干扰,所以通过将反射电极451与第三发光层454r的光程调节为(2n
r-1)λr/4(注意,nr是1以上的自然数)。通过调节光程,可以使第一反射光与第一入射光
的相位一致,从而可以放大来自第三发光层454r的发光。
[0407]
另外,严格而言,可以将反射电极451与第三发光层454r之间的光程称为反射电极451中的反射区域与第三发光层454r中的发光区域之间的光程。但是,难以严格地决定反射电极451中的反射区域或第三发光层454r中的发光区域的位置,所以通过假定反射电极451中的任意的位置为反射区域且假定第三发光层454r中的任意的位置为发光区,来可以充分地获得上述效果。
[0408]
接着,在第二发光元件450g中,因为来自第二发光层454g的发光中的由反射电极451反射而回来的光(第二反射光)与从第二发光层454g直接入射到半透射半反射电极452的光(第二入射光)发生干扰,所以通过将反射电极451与第二发光层454g的光程调节为(2n
g-1)λg/4(注意,ng是1以上的自然数)。通过调节光程,可以使第二反射光与第二入射光的相位一致,从而可以放大来自第二发光层454g的发光。
[0409]
另外,严格而言,可以将反射电极451与第二发光层454g之间的光程称为反射电极451中的反射区域与第二发光层454g中的发光区域之间的光程。但是,难以严格地决定反射电极451中的反射区域或第二发光层454g中的发光区域的位置,所以通过假定反射电极451中的任意的位置为反射区域且假定第二发光层454g中的任意的位置为发光区,来可以充分地获得上述效果。
[0410]
接着,在第三发光元件450b中,因为来自第一发光层454b的发光中的由反射电极451反射而回来的光(第三反射光)与从第一发光层454b直接入射到半透射半反射电极452的光(第三入射光)发生干扰,所以通过将反射电极451与第一发光层454b的光程调节为(2n
b-1)λb/4(注意,nb是1以上的自然数)。通过调节光程,可以使第三反射光与第三入射光的相位一致,从而可以放大来自第一发光层454b的发光。
[0411]
另外,严格而言,可以将反射电极451与第一发光层454b之间的光程称为反射电极451中的反射区域与第一发光层454b中的发光区域之间的光程。但是,难以严格地决定反射电极451中的反射区域或第一发光层454b中的发光区域的位置,所以通过假定反射电极451中的任意的位置为反射区域且假定第一发光层454b中的任意的位置为发光区,来可以充分地获得上述效果。
[0412]
另外,在上述结构中,示出每个发光元件具有在el层中包括多个发光层的结构,但是本发明不局限于此。例如,也可以采用如下结构:将上述结构与实施方式5所说明的串联型(叠层型)发光元件组合,隔着电荷产生层在一个发光元件中设置多个el层,且在各el层中形成一个或多个发光层。
[0413]
本实施方式所示的发光装置具有微腔结构,即使具有相同的el层,也能够提取根据发光元件不同的波长的光,因此不需要rgb的分别涂敷。从而,根据容易实现高精细化等的理由,从实现全彩色化的角度来看上述结构是有利的。另外,因为能够加强特定波长的正面方向的发光强度,所以可以实现低耗电量化。该结构是当将其应用于使用三种颜色以上的像素的彩色显示器(图像显示装置)时特别有效的,但是也可以将其用于照明等的用途。
[0414]
另外,本实施方式所示的结构可以与其他实施方式所示的结构适当地组合而实施。
[0415]
实施方式7
[0416]
在本实施方式中,参照图6a至图7b说明具有本发明的一个方式的发光元件的发光
装置。首先说明图6a和图6b所示的发光装置,接着说明图7a和图7b所示的发光装置。
[0417]
另外,图6a是发光装置的俯视图,图6b是沿着图6a的a-b线和c-d线截断的截面图。
[0418]
在图6a中,以虚线表示的附图标记401、402及403分别表示驱动电路部(源极侧驱动电路)、像素部及指驱动电路部(栅极侧驱动电路)。附图标记404指密封基板,附图标记405指密封材料,被密封材料405包围的部分是空间。
[0419]
注意,引线408是用来传输被输入到源极侧驱动电路401及栅极侧驱动电路403的信号的布线,并接收来自用作外部输入端子的fpc(柔性印刷电路)409的图像信号、时钟信号、起始信号、复位信号等。在此,尽管只显示了fpc,但是该fpc也可以安装有印刷线路板(pwb)。本说明书中的发光装置不仅包括发光装置主体,而且还包括安装有fpc或pwb的发光装置。
[0420]
接着,参照图6b说明截面结构。在元件衬底410上形成有驱动电路部及像素部,这里示出了作为驱动电路部的源极侧驱动电路401和像素部402中的一个像素。
[0421]
另外,在源极侧驱动电路401中形成有组合n沟道型tft423和p沟道型tft424而成的cmos电路。此外,驱动电路也可以利用使用tft形成的各种cmos电路、pmos电路或nmos电路形成。虽然在本实施方式中描述在衬底上形成有驱动电路的驱动器一体型,但是本发明不一定需要限于这种类型,可以不在衬底上,而在外部形成驱动电路。
[0422]
此外,像素部402由多个像素形成,该像素具有开关tft411、电流控制tft412和与电流控制tft412的漏极电连接的第一电极413。注意,以覆盖第一电极413的端部的方式形成有绝缘体414。在此,使用正型光敏丙烯酸树脂膜形成绝缘体414。
[0423]
为了改进覆盖性,在绝缘体414的上端部或下端部形成具有曲率的曲面。例如,当使用正型光敏丙烯酸树脂作为绝缘体414的材料时,优选仅使绝缘体414的上端部具有曲率半径(0.2μm至3μm)的曲面。此外,作为绝缘体414可以使用在受到光照射后不溶解于蚀刻剂的负型或在受到光照射后溶解于蚀刻剂的正型中的任一种。
[0424]
在第一电极413上形成有发光层416和第二电极417。在此,作为用于用作阳极的第一电极413的材料优选使用功函数高的材料。例如,除了ito膜、包含硅的氧化铟锡膜、包含2wt%至20wt%的氧化锌的氧化铟膜、氮化钛膜、铬膜、钨膜、zn膜、pt膜等的单层膜之外,还可以使用氮化钛膜和以铝为主要成分的膜的叠层或氮化钛膜、以铝为主要成分的膜和氮化钛膜的三层结构等。另外,当采用叠层结构时,作为布线的电阻也低,可以得到良好的欧姆接触。
[0425]
此外,可采用各种方法形成发光层416,例如使用蒸镀掩模的蒸镀法、如喷墨法的液滴喷出法、印刷法和旋涂法等。发光层416含有上述实施方式所述的包含嘧啶骨架的有机化合物。此外,发光层416也可以包含另一种材料,例如低分子材料、低聚物、树枝状聚合物或高分子材料。
[0426]
再者,作为用于在发光层416上形成且用作阴极的第二电极417的材料,优选使用具有低功函的材料(例如,al、mg、li、ca或它们的合金或化合物,如mg-ag、mg-in或al-li)。为了使在发光层416中生成的光透过第二电极417,作为第二电极417优选使用具有厚度薄的金属薄膜和透明导电膜(例如,ito、包含2wt%至20wt%氧化锌的氧化铟、包含硅或氧化硅的氧化铟-氧化锡、或氧化锌等)的叠层。
[0427]
再者,通过使用密封材料405将密封衬底404和元件衬底410贴合,在被元件衬底
410、密封衬底404和密封材料405围绕的空间407中设置发光元件418。在空间407中填充有填料,并且除了填充有惰性气体(如氮气或氩气等)之外,有时填充有密封材料405。
[0428]
优选将环氧类树脂用作密封材料405。此外,这些材料优选是尽量不使水分或氧透过的材料。此外,除了玻璃衬底、石英衬底之外,还可以使用frp(fiberglass-reinforced plastics:玻璃纤维增强的塑料)、pvf(polyvinyl fluoride:聚氟乙烯)、聚酯、丙烯酸树脂等构成的塑料衬底作为密封衬底404的材料。
[0429]
如上所述,可以得到具有本发明的一个方式的发光元件的主动矩阵型发光装置。
[0430]
另外,本发明的发光元件可以用于被动矩阵型发光装置,而不局限于上述主动矩阵型发光装置。
[0431]
图7a和图7b示出使用本发明的发光元件的被动矩阵型发光装置的透视图和截面图。注意,图7a是发光装置的透视图,图7b是沿着图7a的x-y线截断的截面图。
[0432]
在图7a和图7b中,在衬底501上的第一电极502和第二电极503之间设置有el层504。第一电极502的端部被绝缘层505覆盖。而且,在绝缘层505上设置有分隔层506。分隔层506的侧壁具有倾斜而使两个侧壁之间的距离向衬底面的方向逐渐变窄。换言之,分隔层506的短边方向上的截面为梯形,并且底边(朝向与绝缘层505的面方向同样的方向且与绝缘层505接触的一边)比上边(朝向与绝缘层505的面方向同样的方向且不与绝缘层505接触的一边)短。像这样,通过设置分隔层506,能够防止因静电等而导致的发光元件的缺陷。
[0433]
如上所述,可以得到具有本发明的一个方式的发光元件的被动矩阵型发光装置。
[0434]
本实施方式中所述的发光装置(主动矩阵型发光装置和被动矩阵型发光装置)都可采用本发明的一个方式中所述的发光元件形成,因此,可以获得低耗电量的发光装置。
[0435]
另外,本实施方式可以与其他实施方式适当地组合。
[0436]
实施方式8
[0437]
在本实施方式中,说明一种电子设备,该电子设备包含上述实施方式所示的本发明的一个方式的发光装置。作为这种电子设备,可以举出影像拍摄装置如摄像机及数码相机等、护目镜型显示器、导航系统、声音再现装置(车载音响、立体声组合音响等)、计算机、游戏机、便携式信息终端(便携式计算机、移动电话、便携式游戏机或电子书阅读器等)、具有记录介质的图像再现装置(具体为再现数字通用光盘(dvd)等记录介质且具有可以显示其图像的显示装置的装置)等。图8a至图8d示出上述电子设备的具体例子。
[0438]
图8a是本发明的一个方式的电视装置,该电视装置包括框体611、支撑台612、显示部613、扬声器部614、视频输入端子615等。在该电视装置中,显示部613可以应用本发明的一个方式的发光装置。因为本发明的一个方式的发光装置可以实现低驱动电压且高电流效率,所以通过应用本发明的一个方式的发光装置,可以获得低耗电量的电视装置。
[0439]
图8b是本发明的一个方式的计算机,该计算机包括主体621、框体622、显示部623、键盘624、外部连接端口625、定点装置626等。在该计算机中,显示部623可以应用本发明的发光装置。因为本发明的发光装置可以实现低驱动电压且高电流效率,所以通过应用本发明的一个方式的发光装置,可以获得低耗电量的计算机。
[0440]
图8c是本发明的一个方式的移动电话,该移动电话包括主体631、框体632、显示部633、声音输入部634、声音输出部635、操作键636、外部连接端口637、天线638等。在该移动电话中,显示部633可以应用本发明的发光装置。因为本发明的发光装置可以实现低驱动电
压且高电流效率,所以通过应用本发明的一个方式的发光装置,可以获得低耗电量的移动电话。
[0441]
图8d是本发明的一个方式的影像拍摄装置,该影像拍摄装置包括主体641、显示部642、框体643、外部连接端口644、遥控接收部645、影像接收部646、电池647、声音输入部648、操作键649、取景部650等。在该影像拍摄装置中,显示部642可以应用本发明的一个方式的发光装置。因为本发明的一个方式的发光装置可以驱动电压低地得到高电流效率,所以通过应用本发明的一个方式的发光装置,可以获得低耗电量的影像拍摄装置。
[0442]
如上所述,本发明的一个方式的发光装置的应用范围非常广泛,并且可以将该发光装置应用于各种领域的电子设备。通过使用本发明的一个方式的发光装置,可以获得减少了耗电量的电子设备。
[0443]
此外,也可以将本发明的一个方式的发光装置用作照明装置。图9a是将本发明的一个方式的发光装置用作背光灯的液晶显示装置的一个例子。图9a所示的液晶显示装置包括框体701、液晶层702、背光灯703以及框体704,液晶层702与驱动器ic705连接。此外,作为背光灯703使用本发明的一个方式的发光装置,通过端子706向背光灯703供应电流。
[0444]
如上所述,通过将本发明的一个方式的发光装置用作液晶显示装置的背光灯,可以获得耗电量低的背光灯。另外,由于本发明的一个方式的发光装置是面发光的照明装置,也可以实现大面积化,所以也可以实现背光灯的大面积化。因此,可以获得实现低耗电量和大面积的液晶显示装置。
[0445]
接着,图9b是将本发明的一个方式的发光装置用作照明装置的台灯的例子。图9b所示的台灯包括框体801和光源802,将本发明的一个方式的发光装置用作光源802。因为可以实现低驱动电压且高电流效率,所以通过应用本发明的一个方式的发光装置,可以获得低耗电量的台灯。
[0446]
接着,图9c是将本发明的一个方式的发光装置用作室内照明装置901的例子。由于本发明的一个方式的发光装置也可以实现大面积化,所以可以用作大面积的照明装置。另外,因为本发明的一个方式的发光装置可以驱动电压低地得到高电流效率,所以通过应用本发明的一个方式的发光装置,可以获得低耗电量的照明装置。像这样,可以在将本发明的一个方式的发光装置用作室内照明装置901的房间内设置如图8a所说明的本发明的一个方式的电视装置902,来看公共广播或电影。
[0447]
另外,本实施方式可以与其他实施方式适当地组合。
[0448]
另外,本发明的一个方式的包含嘧啶骨架的有机化合物(第二化合物)可以用于有机薄膜太阳能电池。具体而言,它具有载流子传输性,因此可以将其用于载流子传输层及载流子注入层。另外,它能够光激发,所以可以将其用于发电层。
[0449]
实施例1
[0450]
在本实施例中,作为第二化合物的包含嘧啶骨架的有机化合物的一个方式,对由下述结构式(300)表示的4,6-双[3-(菲-9-基)苯基]嘧啶(简称:4,6mpnp2pm)的合成方法进行说明。
[0451][0452]
《4,6-双[3-(菲-9-基)苯基]嘧啶(简称:4,6mpnp2pm)的合成》
[0453]
(c-1)示出4,6-双[3-(菲-9-基)苯基]嘧啶(简称:4,6mpnp2pm)的合成方案。
[0454][0455]
在100ml圆底烧瓶中,放入0.64g(4.3mmol)4,6-二氯嘧啶、3.2g(11mmol)3-(菲-9-基)苯基硼酸及2.3g(21mmol)碳酸钠,对该混合物加入10ml乙腈及20ml水。边降低压力边搅拌该混合物,来进行脱气。对该混合物加入30mg(43μmol)双(三苯基膦)二氯化钯(i i)。在氩气氛下照射1.5小时的微波(2.45ghz,100w)来进行加热,搅拌。对该混合物加入1.0g(3.4mmol)3-(9-菲基)苯基硼酸、0.71g(6.7mmol)碳酸钠、28mg(40μmol)双(三苯基膦)二氯化钯(i i),再加热1小时,搅拌。加热后,对该混合物加入水,用二氯甲烷萃取有机层。用饱和碳酸氢钠水溶液、饱和食盐水洗涤得到的有机层,加入硫酸镁来吸附水分。对该混合物进行自然过滤,浓缩滤液来得到固体。利用硅胶柱色谱法使该固体纯化。在硅胶柱色谱法中,使用甲苯作为展开剂,浓缩所得到的馏分得到固体。使用甲苯/己烷使其再结晶,以40%的收率得到4,6-双[3-(菲-9-基)苯基]嘧啶(简称:4,6mpnp2pm)的白色固体0.99g。
[0456]
通过利用梯度升华法(train sublimation method)纯化所得到的4,6-双[3-(菲-9-基)苯基]嘧啶(简称:4,6mpnp2pm)的白色固体。在压力为3.7pa且氩流量为5.0ml/min的条件下,以280℃对4,6-双[3-(菲-9-基)苯基]嘧啶(简称:4,6mpnp2pm)进行14小时的加热来进行纯化。纯化之后,以79%的收率得到4,6-双[3-(菲-9-基)苯基]嘧啶(简称:4,
6mpnp2pm)的白色固体0.80g。
[0457]
通过核磁共振法(1h nmr)确认到上述化合物是目的物的4,6-双[3-(菲-9-基)苯基]嘧啶(简称:4,6mpnp2pm)。
[0458]
以下示出所得到的化合物的1h nmr数据。
[0459]1h nmr(cdcl3,300mhz):δ=7.50-7.75(m,14h),7.87-7.91(m,4h),8.21-8.31(m,5h),8.73(d,j1=8.4hz,2h),8.78(d,j1=8.4hz,2h),9.36(d,j1=0.9hz,1h)。
[0460]
另外,图10a和图10b示出1h nmr图。另外,图10b为放大图10a的7.0ppm至9.5ppm的范围而示出的图表。
[0461]
接着,通过液相色谱-质谱联用分析(liquid chromatography mass spectrometry:lc/ms分析)对在本实施例中得到的4,6-双[3-(菲-9-基)苯基]嘧啶(简称:4,6mpnp2pm)进行分析。
[0462]
在lc/ms分析中,利用沃特世(waters)公司制造的acquity uplc进行lc(液相色谱)分离,并利用沃特世公司制造的xevo g2 tof ms进行ms分析(质量分析)。lc分离中使用的色谱柱为acquity uplc beh c8(2.1
×
100mm,1.7μm),色谱柱温度为40℃。作为流动相a使用乙腈,作为流动相b使用0.1%甲酸水溶液。另外,以任意浓度将4,6mpnp2pm溶解于甲苯中并利用乙腈稀释来调节作为样品,注入量为5.0μl。
[0463]
在lc分离中利用改变流动相的组成的梯度法,检测开始后0分钟至1分钟的比为流动相a:流动相b=40:60,然后以检测开始后10分钟的比为流动相a:流动相b=95:5的方式改变组成。线性地改变组成比。
[0464]
在ms分析中利用电喷雾电离法(electrospray ionization:esi)进行离子化,毛细管电压为3.0kv,样品锥孔电压为30v,利用正模式进行检测。另外,所检测的质量范围为m/z=100至1200。
[0465]
在碰撞室(collision cell)中使在如上条件下分离并离子化了的m/z=584.23的成分碰撞到氩气来解离为产物离子。碰撞氩时的能量(碰撞能量:collision energy)为70ev。图97示出利用飞行时间质谱(tof-ms)对被解离的产物离子进行ms分析的结果。
[0466]
由图97的结果可知,在本发明的一个方式的4,6mpnp2pm中,主要在m/z=252附近、m/z=277附近及m/z=541附近检测出部分骨架的产物离子的峰值,主要在m/z=585附近检测出来源于前体离子的峰值,主要在m/z=1169附近检测出来源于二聚离子的峰值。另外,图97的结果示出来源于4,6mpnp2pm的特征,由此可以说是为了识别混合物所包含的4,6mpnp2pm而重要的数据。
[0467]
另外,位于m/z=541附近的碎片可以认为是因4,6mpnp2pm中的嘧啶环开环而产生的产物离子,并且这种产物离子的形式是在嘧啶环的4位及6位被取代的本发明的一个方式的有机化合物的特征之一。从而,这表示本发明的一个方式的4,6mpnp2pm包含在4位及6位被取代的嘧啶环。
[0468]
另外,图98a至图98d示出利用飞行时间二次离子质谱分析仪(time-of-flight secondary ion mass spectrometer:tof-sims)测定的4,6mpnp2pm的定性光谱(正离子及负离子)。
[0469]
另外,图98a示出正离子的测定结果,横轴表示0至500范围的m/z,纵轴表示强度(任意单位)。另外,图98b示出正离子的测定结果,横轴表示400至1200范围的m/z,纵轴表示
强度(任意单位)。另外,图98c示出负离子的测定结果,横轴表示0至500范围的m/z,纵轴表示强度(任意单位)。另外,图98d示出负离子的测定结果,横轴表示400至1200范围的m/z,纵轴表示强度(任意单位)。
[0470]
作为装置使用tof sims5(ion-tof公司制造),作为一次离子源使用bi
3
。以脉冲宽度为7至12nm的脉冲状的方式照射一次离子,照射量为8.2
×
10
10
至6.7
×
10
11
ions/cm2(1
×
10
12
ions/cm2以下),加速电压为25kev,电流值为0.2pa。另外,作为样品使用4,6mpnp2pm的粉末进行测定。
[0471]
由图98a和图98b的结果可知,在本发明的一个方式的4,6mpnp2pm中,主要在m/z=252附近及m/z=276附近检测出部分骨架的产物离子的峰值。
[0472]
另外,由图98c和图98d的结果可知,在本发明的一个方式的4,6mpnp2pm中,主要在m/z=571附近检测出部分骨架的产物离子的峰值,主要在m/z=585及m/z=595附近检测出来源于前体离子的峰值。另外,图98c和图98d的结果示出来源于4,6mpnp2pm的特征,由此可以说这是为了识别混合物所包含的4,6mpnp2pm而重要的数据。
[0473]
此外,图11a示出4,6mpnp2pm的甲苯溶液的吸收光谱,图11b示出其发射光谱。另外,图12a示出4,6mpnp2pm的薄膜的吸收光谱,图12b示出其发射光谱。使用紫外可见分光光度计(日本分光株式会社制造,v550型)进行吸收光谱的测量。将溶液放在石英皿中,并在石英衬底上蒸镀薄膜制造样品来进行测量。通过减去仅将甲苯放在石英皿中而测量出来的吸收光谱,获得该溶液的吸收光谱。通过减去石英衬底的吸收光谱,获得薄膜的吸收光谱。在图11a至图12b中,横轴表示波长(nm),纵轴表示强度(任意单位)。在测量甲苯溶液的情况下,在281nm附近及299nm附近附近观察到吸收峰值,在322nm附近、342nm附近及357nm附近附近观察到发光波长的峰值。另外,在测量薄膜的情况下,在206nm附近、257nm附近、304nm附近及353nm附近观察到吸收峰值,在406nm附近观察到发光波长的峰值。
[0474]
另外,利用差示扫描量热仪(differential scan calorimeter:dsc)测定在本实施例中合成的4,6mpnp2pm的玻璃化转变温度。测定结果示出4,6mpnp2pm的玻璃化转变温度为126℃。如此,4,6mpnp2pm具有高玻璃化转变温度,所以可知4,6mpnp2pm具有良好的耐热性。另外,观察不到4,6mpnp2pm的结晶峰,所以可知4,6mpnp2pm是不容易晶化的物质。
[0475]
另外,测定4,6mpnp2pm的电化学特性(薄膜)(测定装置:日本理研记器株式会社制造,ac-2)。另外,电化学特性(薄膜)的测定如下所述进行。
[0476]
将在空气中用光电子分光光度计(日本理研计器株式会社制造,ac-2)测得的电离电势值转化为负值,获得homo能级值。按照以下方式获得lumo能级值:假设直接跃迁,使用本实施例所述的薄膜的吸收光谱上的数据,由tauc曲线获得吸收边缘,该吸收边缘被看做是光学能隙并加到homo能级值上。
[0477]
由电化学特性(薄膜)的测定结果可知,4,6mpnp2pm的homo能级(最高占有分子轨道)为-5.95ev,lumo能级(最低未占分子轨道)为-2.70ev,并且能隙(bg)为3.25ev。
[0478]
由以上结果可知4,6mpnp2pm具有深homo能级、浅lumo能级及宽能隙(bg)。
[0479]
实施例2
[0480]
在本实施例中,对由下述结构式(311)表示的4,6-双[3-(萘-1-基)苯基]-2-苯基嘧啶(简称:2ph-4,6mnp2pm)的合成方法进行说明。
[0481][0482]
《4,6-双[3-(萘-1-基)苯基]-2-苯基嘧啶(简称:2ph-4,6mnp2pm)的合成》
[0483]
(d-1)示出4,6-双[3-(萘-1-基)苯基]-2-苯基嘧啶(简称:2ph-4,6mnp2pm)的合成方案。
[0484][0485]
在100ml三口烧瓶中,放入1.80g(3.86mmol)4,6-双(3-溴苯基)-2-苯基嘧啶、4.50g(9.65mmol)1-萘硼酸及117mg(386μmol)三(2-甲基苯基)膦,对该混合物加入10ml 2.0m碳酸钾水溶液、15ml甲苯及5ml乙醇,并且在减压下搅拌而进行脱气。对该混合物加入17mg(77.2μmol)乙酸钯(ii),在氮气流下以90℃搅拌8小时。搅拌后,过滤所得到的混合物,回收固体。接着,用甲苯萃取滤液的水层。得到的萃取溶液和有机层合并用饱和食盐水洗涤,加入硫酸镁来吸附水分。对该混合物进行自然过滤,浓缩滤液来得到固体。在该固体中加入甲苯,通过硅藻土(日本和光纯药工业株式会社、目录号码:531-16855)、矾土、硅酸镁(日本和光纯药工业株式会社、目录号码:540-00135)抽滤,浓缩滤液来得到固体。利用硅胶柱色谱法(作为展开剂使用己烷,然后使用甲苯:己烷=1:4)使该固体纯化。使用甲苯使得到的固体再结晶,以85%的收率得到白色固体1.84g。
[0486]
通过利用梯度升华法纯化所得到的白色固体1.66g。在压力为2.9pa且氩流量为5ml/min的条件下,以260℃进行加热来纯化。纯化之后,以92%的收率得到白色固体1.52g。
[0487]
通过核磁共振法(1h nmr)确认到上述化合物是目的物的4,6-双[3-(萘-1-基)苯基]-2-苯基嘧啶(简称:2ph-4,6mnp2pm)。
[0488]
以下示出所得到的化合物的1h nmr数据。
[0489]1h nmr(cdcl3,300mhz):δ=7.42-7.60(m,11h),7.65-7.72(m,4h),7.90-7.96(m,6h),8.10(s,1h),8.36-8.40(m,4h),8.69-8.73(m,2h)。
[0490]
另外,图13a和图13b示出1h nmr图。另外,图13b为放大图13a的7.0ppm至9.5ppm的范围而示出的图表。
[0491]
接着,通过液相色谱-质谱联用分析(lc/ms分析)对在本实施例中得到的4,6-双[3-(萘-1-基)苯基]-2-苯基嘧啶(简称:2ph-4,6mnp2pm)进行分析。
[0492]
在lc/ms分析中,利用沃特世(waters)公司制造的acquity uplc进行lc(液相色谱)分离,并利用沃特世公司制造的xevo g2 tof ms进行ms分析(质量分析)。lc分离中使用的色谱柱为acquity uplc beh c8(2.1
×
100mm,1.7μm),色谱柱温度为40℃。作为流动相a使用乙腈,作为流动相b使用0.1%甲酸水溶液。另外,以任意浓度将2ph-4,6mnp2pm溶解于甲苯中并利用乙腈稀释来调节作为样品,注入量为5.0μl。
[0493]
在lc分离中利用改变流动相的组成的梯度法,检测开始后0分钟至1分钟的比为流动相a:流动相b=40:60,然后以检测开始后10分钟的比为流动相a:流动相b=95:5的方式改变组成。线性地改变组成比。
[0494]
在ms分析中利用电喷雾电离法(esi)进行离子化,毛细管电压为3.0kv,样品锥孔电压为30v,利用正模式进行检测。另外,所检测的质量范围为m/z=100至1200。
[0495]
在碰撞室(collision cell)中使在如上条件下分离并离子化了的m/z=560.23的成分碰撞到氩气来解离为产物离子。碰撞氩时的能量(碰撞能量:collision energy)为70ev。图99示出利用飞行时间质谱(tof-ms)对被解离的产物离子进行ms分析的结果。
[0496]
由图99的结果可知,在本发明的一个方式的2ph-4,6mnp2pm中,主要在m/z=202附近、m/z=230附近m/z=253附近及m/z=441附近检测出部分骨架的产物离子的峰值,主要在m/z=561附近检测出来源于前体离子的峰值,主要在m/z=1121附近检测出来源于二聚离子的峰值。另外,图99的结果示出来源于2ph-4,6mnp2pm的特征,由此可以说是为了识别混合物所包含的2ph-4,6mnp2pm而重要的数据。
[0497]
另外,位于m/z=441附近的碎片可以认为是因2ph-4,6mnp2pm中的嘧啶环开环(同时失去2位的苯基)而产生的产物离子,并且这种产物离子的形式是在嘧啶环的4位及6位被取代的本发明的一个方式的有机化合物的特征之一。从而,这表示本发明的一个方式的2ph-4,6mnp2pm包含在4位及6位被取代的嘧啶环。
[0498]
此外,图14a示出2ph-4,6mnp2pm的甲苯溶液的吸收光谱,图14b示出其发射光谱。另外,图15a示出2ph-4,6mnp2pm的薄膜的吸收光谱,图15b示出其发射光谱。使用紫外可见分光光度计(日本分光株式会社制造,v550型)进行吸收光谱的测量。将溶液放在石英皿中,并在石英衬底上蒸镀薄膜制造样品来进行测量。通过减去仅将甲苯放在石英皿中而测量出来的吸收光谱,获得该溶液的吸收光谱。通过减去石英衬底的吸收光谱,获得薄膜的吸收光谱。在图14a至图15b中,横轴表示波长(nm),纵轴表示强度(任意单位)。在测量甲苯溶液的情况下,在282nm附近观察到吸收峰值,在361nm附近观察到发光波长的峰值。另外,在测量薄膜的情况下,在228nm附近、266nm附近、279nm附近、316nm附近及357nm附近观察到吸收峰值,在393nm附近观察到发光波长的峰值。
[0499]
实施例3
[0500]
在本实施例中,对由下述结构式(314)表示的4,6-双[3-(三亚苯基-2-基)苯基]嘧
啶(简称:4,6mtpp2pm)的合成方法进行说明。
[0501][0502]
《4,6-双[3-(三亚苯基-2-基)苯基]嘧啶(简称:4,6mtpp2pm)的合成》
[0503]
(e-1)示出4,6-双[3-(三亚苯基-2-基)苯基]嘧啶(简称:4,6mtpp2pm)的合成方案。
[0504][0505]
在100ml圆底烧瓶中,放入0.65g(4.38mmol)4,6-二氯嘧啶、3.83g(11.0mmol)3-(三亚苯基-2-基)苯基硼酸及2.33g(22.0mol)碳酸钠,对该混合物加入20ml 1,3-二甲基-3,4,5,6-四氢-2(1h)-嘧啶酮(简称:dmpu)及10ml水。边降低压力边搅拌该混合物,来进行脱气。对该混合物加入41mg(52.6μmol)双(三苯基膦)钯(i i)二氯化物,用氩气置换空气。在氩气流下对该反应容器照射1.5小时的微波(2.45ghz,100w)来进行加热。加热后,对该混合物加入水并过滤,以得到滤渣。使用乙醇对所得到的固体进行洗涤。在该固体中加入甲苯,通过硅藻土(日本和光纯药工业株式会社、目录号码:531-16855)、矾土、硅酸镁(日本和光纯药工业株式会社、目录号码:540-00135)抽滤,浓缩滤液来得到白色固体。利用硅胶柱色谱法(作为展开剂使用甲苯,然后使用甲苯:乙酸乙酯=10:1)使该固体纯化。使用甲苯使得到的固体再结晶,以37%的收率得到白色固体1.12g。
[0506]
通过利用梯度升华法纯化所得到的白色固体0.88g。在压力为2.8pa且氩流量为
5ml/min的条件下,以370℃进行加热来纯化。纯化之后,以81%的收率得到淡黄色固体0.71g。
[0507]
通过核磁共振法(1h nmr)确认到上述化合物是目的物的4,6-双[3-(三亚苯基-2-基)苯基]嘧啶(简称:4,6mtpp2pm)。
[0508]
以下示出所得到的物质的1h nmr数据。
[0509]1h nmr(cdcl3,300mhz):δ=7.66-7.76(m,10h),7.98-8.02(m,4h),8.23(d,j1=7.8hz,2h),8.34(s,1h),8.62-8.81(m,12h),8.95(s,2h),9.46(s,1h)。
[0510]
另外,图16a和图16b示出1h nmr图。另外,图16b为放大图16a的7.0ppm至9.5ppm的范围而示出的图表。
[0511]
接着,通过液相色谱-质谱联用分析(lc/ms分析)对在本实施例中得到的4,6-双[3-(三亚苯基-2-基)苯基]嘧啶(简称:4,6mtpp2pm)进行分析。
[0512]
在lc/ms分析中,利用沃特世(waters)公司制造的acquity uplc进行lc(液相色谱)分离,并利用沃特世公司制造的xevo g2 tof ms进行ms分析(质量分析)。lc分离中使用的色谱柱为acquity uplc beh c8(2.1
×
100mm,1.7μm),色谱柱温度为40℃。作为流动相a使用乙腈,作为流动相b使用0.1%甲酸水溶液。另外,以任意浓度将4,6mtpp2pm溶解于氯仿中并利用乙腈稀释来调节作为样品,注入量为5.0μl。
[0513]
在lc分离中利用改变流动相的组成的梯度法,检测开始后0分钟至1分钟的比为流动相a:流动相b=40:60,然后以检测开始后10分钟的比为流动相a:流动相b=95:5的方式改变组成。线性地改变组成比。
[0514]
在ms分析中利用电喷雾电离法(esi)进行离子化,毛细管电压为3.0kv,样品锥孔电压为30v,利用正模式进行检测。另外,所检测的质量范围为m/z=100至1200。
[0515]
在碰撞室(collision cell)中使在如上条件下分离并离子化了的m/z=684.26的成分碰撞到氩气来解离为产物离子。碰撞氩时的能量(碰撞能量:collision energy)为70ev。图100示出利用飞行时间质谱(tof-ms)对被解离的产物离子进行ms分析的结果。
[0516]
由图100的结果可知,在本发明的一个方式的4,6mtpp2pm中,主要在m/z=327附近及m/z=641附近检测出部分骨架的产物离子的峰值,主要在m/z=685附近检测出前体离子。另外,图100的结果示出来源于4,6mtpp2pm的特征,由此可以说是为了识别混合物所包含的4,6mtpp2pm而重要的数据。
[0517]
另外,位于m/z=641附近的产物离子可以认为是因4,6mtpp2pm中的嘧啶环开环而产生的产物离子,并且这种产物离子的形式是在嘧啶环的4位及6位被取代的本发明的一个方式的有机化合物的特征之一。从而,这表示本发明的一个方式的4,6mtpp2pm包含在4位及6位被取代的嘧啶环。
[0518]
此外,图17a示出4,6mtpp2pm的甲苯溶液的吸收光谱,图17b示出其发射光谱。另外,图18a示出4,6mtpp2pm的薄膜的吸收光谱,图18b示出其发射光谱。使用紫外可见分光光度计(日本分光株式会社制造,v550型)进行吸收光谱的测量。将溶液放在石英皿中,并在石英衬底上蒸镀薄膜制造样品来进行测量。通过减去仅将甲苯放在石英皿中而测量出来的吸收光谱,获得该溶液的吸收光谱。通过减去石英衬底的吸收光谱,获得薄膜的吸收光谱。在图17a至图18b中,横轴表示波长(nm),纵轴表示强度(任意单位)。在测量甲苯溶液的情况下,在281nm附近观察到吸收峰值,在363nm附近观察到发光波长的峰值。另外,在测量薄膜
的情况下,在271nm附近及320nm附近观察到吸收峰值,在423nm附近观察到发光波长的峰值。
[0519]
实施例4
[0520]
在本实施例中,对由下述结构式(400)表示的4,6-双[3-(二苯并噻吩-4-基)苯基]嘧啶(简称:4,6mdbtp2pm-ii)的合成方法进行说明。
[0521][0522]
《4,6-双[3-(二苯并噻吩-4-基)苯基]嘧啶(简称:4,6mdbtp2pm-ii)的合成》
[0523]
(f-1)示出4,6-双[3-(二苯并噻吩-4-基)苯基]嘧啶(简称:4,6mdbtp2pm-ii)的合成方案。
[0524][0525]
在100ml茄形烧瓶中,放入1.0g(6.7mmol)4,6-二氯嘧啶、5.1g(17mmol)3-(二苯并噻吩-4-基)-苯基硼酸及3.5g(34mmol)碳酸钠、20ml1,3-二甲基-3,4,5,6-四氢-2(1h)-嘧啶酮(dmpu)及10ml水。边降低压力边搅拌该混合物,来进行脱气。对该混合物加入56mg(81μmol)双(三苯基膦)二氯化钯(ii),用氩气置换空气。对反应容器照射1.5小时的微波(2.45ghz,100w)来进行加热,搅拌。加热后,对该混合物加入水,过滤,得到滤渣。用二氯甲烷和乙醇洗涤得到的固体。在得到的固体中加入甲苯,通过硅藻土(日本和光纯药工业株式会社、目录号码:531-16855)、矾土、硅酸镁(日本和光纯药工业株式会社、目录号码:540-00135)抽滤,浓缩滤液来得到固体。使用甲苯使得到的固体再结晶,以63%的收率得到白色固体2.52g。
[0526]
通过利用梯度升华法纯化所得到的白色固体2.50g。在压力为3.6pa且氩流量为5ml/min的条件下,以300℃进行加热来纯化。纯化之后,以79%的收率得到白色固体1.98g。
[0527]
通过核磁共振法(1h nmr)确认到上述化合物是目的物的4,6-双[3-(二苯并噻吩-4-基)苯基]嘧啶(简称:4,6mdbtp2pm-i i)。
[0528]
以下示出所得到的化合物的1h nmr数据。
[0529]1h nmr(cdcl3,300mhz):δ=7.41-7.51(m,4h),7.58-7.62(m,4h),7.68-7.79(m,4h),8.73(dt,j1=8.4hz,j2=0.9hz,2h),8.18-8.27(m,7h),8.54(t,j1=1.5hz,2h),9.39(d,j1=0.9hz,1h)。
[0530]
另外,图19a和图19b示出1h nmr图。另外,图19b为放大图19a的7.0ppm至9.5ppm的范围而示出的图表。
[0531]
接着,通过液相色谱-质谱联用分析(lc/ms分析)对在本实施例中得到的4,6-双[3-(二苯并噻吩-4-基)苯基]嘧啶(简称:4,6mdbtp2pm-ii)进行分析。
[0532]
在lc/ms分析中,利用沃特世(waters)公司制造的acquity uplc进行lc(液相色谱)分离,并利用沃特世公司制造的xevo g2 tof ms进行ms分析(质量分析)。lc分离中使用的色谱柱为acquity uplc beh c8(2.1
×
100mm,1.7μm),色谱柱温度为40℃。作为流动相a使用乙腈,作为流动相b使用0.1%甲酸水溶液。另外,另外,以任意浓度将4,6mdbtp2pm-ii溶解于氯仿中并利用乙腈稀释来调节作为样品,注入量为5.0μl。
[0533]
在lc分离中利用改变流动相的组成的梯度法,检测开始后0分钟至1分钟的比为流动相a:流动相b=40:60,然后以检测开始后10分钟的比为流动相a:流动相b=95:5的方式改变组成。线性地改变组成比。
[0534]
在ms分析中利用电喷雾电离法(esi)进行离子化,毛细管电压为3.0kv,样品锥孔电压为30v,利用正模式进行检测。另外,所检测的质量范围为m/z=100至1200。
[0535]
在碰撞室(collision cell)中使在如上条件下分离并离子化了的m/z=596.14的成分碰撞到氩气来解离为产物离子。碰撞氩时的能量(碰撞能量:collision energy)为70ev。图101示出利用飞行时间质谱(tof-ms)对被解离的产物离子进行ms分析的结果。
[0536]
由图101的结果可知,在本发明的一个方式的4,6mdbtp2pm-ii中,主要在m/z=252附近、m/z=258附近、m/z=284附近、m/z=309附近及m/z=553附近检测出部分骨架的产物离子的峰值,主要在m/z=597附近检测出来源于前体离子的峰值,主要在m/z=1193附近检测出来源于二聚离子的峰值。另外,图101的结果示出来源于4,6mdbtp2pm-ii的特征,由此可以说是为了识别混合物所包含的4,6mdbtp2pm-ii而重要的数据。
[0537]
另外,位于m/z=553附近的产物离子可以认为是因4,6mdbtp2pm-ii中的嘧啶环开环而产生的碎片,并且这种产物离子的形式是在嘧啶环的4位或6位被取代的本发明的一个方式的有机化合物的特征之一。从而,这表示本发明的一个方式的4,6mdbtp2pm-ii包含在4位或6位被取代的嘧啶环。
[0538]
另外,图102a至图102d示出利用飞行时间二次离子质谱分析仪(tof-sims)测定的4,6mdbtp2pm-ii的定性光谱(正离子及负离子)。
[0539]
另外,图102a示出正离子的测定结果,横轴表示0至500范围的m/z,纵轴表示强度(任意单位)。另外,图102b示出正离子的测定结果,横轴表示400至1200范围的m/z,纵轴表示强度(任意单位)。另外,图102c示出负离子的测定结果,横轴表示0至500范围的m/z,纵轴表示强度(任意单位)。另外,图102d示出负离子的测定结果,横轴表示400至1200范围的m/z,纵轴表示强度(任意单位)。
[0540]
作为装置使用tof sims5(ion-tof公司制造),作为一次离子源使用bi
3
。以脉冲宽度为7至12nm的脉冲状的方式照射一次离子,照射量为8.2
×
10
10
至6.7
×
10
11
ions/cm2(1
×
10
12
ions/cm2以下),加速电压为25kev,电流值为0.2pa。另外,作为样品使用4,6mdbtp2pm-ii的粉末进行测定。
[0541]
由图102a和图102b的结果可知,在本发明的一个方式的4,6mdbtp2pm-ii中,主要在m/z=184附近、m/z=258附近、m/z=271附近、m/z=284附近、m/z=296附近、m/z=309及m/z=597附近检测出部分骨架的产物离子的峰值。
[0542]
另外,由图102c和图102d的结果可知,在本发明的一个方式的4,6mdbtp2pm-ii中,主要在m/z=583附近检测出部分骨架的产物离子的峰值,主要在m/z=597附近、m/z=607附近及m/z=627附近检测出来源于前体离子的峰值。另外,图102c和图102d的结果示出来源于4,6mdbtp2pm-ii的特征,由此可以说这是为了识别混合物所包含的4,6mdbtp2pm-ii而重要的数据。
[0543]
另外,利用gc/ms仪器(赛默飞世尔科技公司(thermo fisher scientific inc.)制造,itq1100离子阱型gc/msn直接进样系统/dep)测定本发明的一个方式的4,6mdbtp2pm-ii。模式为ei ,离子化电压值为70ev,发射电流值为250μa,电子透镜设置为15v。另外,以升温速度10℃/sec的方式将样品温度提高到1000℃。从测定结果可知,在本发明的一个方式的4,6mdbtp2pm-ii(m/z=596.14)中,主要在m/z=184附近、m/z=282附近、m/z=298附近、m/z=310附近、m/z=552附近及m/z=568附近检测出部分骨架的产物离子的峰值,主要在m/z=596附近检测出来源于前体离子的峰值。另外,这些结果示出来源于4,6mdbtp2pm-ii的特征,由此可以说这是为了识别混合物所包含的4,6mdbtp2pm-ii而重要的数据。
[0544]
另外,位于m/z=184附近的产物离子可以认为是来源于4,6mdbtp2pm-ii中的二苯并噻吩的碎片,并且这种产物离子的形式是具有二苯并噻吩骨架的本发明的一个方式的有机化合物的特征之一。从而,这表示本发明的一个方式的4,6mdbtp2pm-ii包含二苯并噻吩骨架。
[0545]
另外,位于m/z=552附近及m/z=568附近的产物离子可以认为是因4,6mdbtp2pm-ii中的嘧啶环开环而产生的碎片,并且这种产物离子的形式是在嘧啶环的4位及6位被取代的本发明的一个方式的杂环化合物的特征之一。从而,这表示本发明的一个方式的4,6mdbtp2pm-ii包含在4位及6位被取代的嘧啶环。
[0546]
此外,图20a示出4,6mdbtp2pm-ii的甲苯溶液的吸收光谱,图20b示出其发射光谱。另外,图21a示出4,6mdbtp2pm-ii的薄膜的吸收光谱,图21b示出其发射光谱。使用紫外可见分光光度计(日本分光株式会社制造,v550型)进行吸收光谱的测量。将溶液放在石英皿中,并在石英衬底上蒸镀薄膜制造样品来进行测量。通过减去仅将甲苯放在石英皿中而测量出来的吸收光谱,获得该溶液的吸收光谱。通过减去石英衬底的吸收光谱,获得薄膜的吸收光谱。
[0547]
在图20a至图21b中,横轴表示波长(nm),纵轴表示强度(任意单位)。在测量甲苯溶液的情况下,在282nm附近观察到吸收峰值,在376nm附近观察到发光波长的峰值。另外,在测量薄膜的情况下,在244nm、265nm、290nm、317nm及334nm附近观察到吸收峰值,在396nm附近观察到发光波长的峰值。
[0548]
实施例5
[0549]
在本实施例中,对由下述结构式(401)表示的2,4-双[3-(二苯并噻吩-4-基)苯基]嘧啶(简称:2,4mdbtp2pm-ii)的合成方法进行说明。
[0550][0551]
《2,4-双[3-(二苯并噻吩-4-基)苯基]嘧啶(简称:2,4mdbtp2pm-ii)的合成》
[0552]
(g-1)示出2,4-双[3-(二苯并噻吩-4-基)苯基]嘧啶(简称:2,4mdbtp2pm-ii)的合成方案。
[0553][0554]
在100ml茄形烧瓶中,放入0.75g(5.03mmol)的2,4-二氯嘧啶、3.82g(12.6mmol)3-(二苯并噻吩-4-基)苯基硼酸及2.67g(25.2mmol)碳酸钠,对该混合物加入20ml 1,3-二甲基-3,4,5,6-四氢-2(1h)-嘧啶酮(简称:dmpu)及10ml水。边降低压力边搅拌该混合物,来进行脱气。对该混合物加入42mg(60.3μmol)双(三苯基膦)钯(ii)二氯化物,用氩气置换空气。在氩气流下对该反应容器照射55分钟的微波(2.45ghz,100w)来进行加热。加热后,对该混合物加入水并过滤,以得到滤渣。使用乙醇及二氯甲烷对所得到的固体进行洗涤。在该固体中加入甲苯,通过硅藻土(日本和光纯药工业株式会社、目录号码:531-16855)、矾土、硅酸镁(日本和光纯药工业株式会社、目录号码:540-00135)抽滤,浓缩滤液来得到白色固体。利用硅胶柱色谱法(作为展开剂使用甲苯)使该固体纯化。使用甲苯使得到的固体再结晶,以62%的收率得到白色固体1.87g。
[0555]
通过利用梯度升华法纯化所得到的固体1.80g。在压力为5.1pa且氩流量为10ml/min的条件下,以300℃进行加热来纯化。纯化之后,以68%的收率得到白色固体1.98g。
[0556]
通过核磁共振法(1h nmr)确认到上述化合物是目的物的2,4-双[3-(二苯并噻吩-4-基)苯基]嘧啶(简称:2,4mdbtp2pm-ii)。
[0557]
以下示出所得到的物质的1h nmr数据。
[0558]1h nmr(cdcl3,300mhz):δ=7.39-7.76(m,13h),7.90(dt,j1=7.8hz,j2=1.5hz,
2h),8.13-8.20(m,4h),8.32(dt,j1=8.1hz,j2=1.5hz,1h),8.68(dt,j1=7.8hz,j2=1.5hz,1h),8.72(t,j1=1.5hz,1h),8.92(d,j1=5.4hz,1h),9.07(t,j1=1.5hz,1h)。
[0559]
另外,图22a和图22b示出1h nmr图。另外,图22b为放大图22a的7.0ppm至9.5ppm的范围而示出的图表。
[0560]
此外,图23a示出2,4mdbtp2pm-ii的甲苯溶液的吸收光谱,图23b示出其发射光谱。另外,图24a示出2,4mdbtp2pm-ii的薄膜的吸收光谱,图24b示出其发射光谱。使用紫外可见分光光度计(日本分光株式会社制造,v550型)进行吸收光谱的测量。将溶液放在石英皿中,并在石英衬底上蒸镀薄膜制造样品来进行测量。通过减去仅将甲苯放在石英皿中而测量出来的吸收光谱,获得该溶液的吸收光谱。通过减去石英衬底的吸收光谱,获得薄膜的吸收光谱。在图23a至图24b中,横轴表示波长(nm),纵轴表示强度(任意单位)。在测量甲苯溶液的情况下,在283nm附近观察到吸收峰值,在356nm附近观察到发光波长的峰值。另外,在测量薄膜的情况下,在244nm附近、226nm附近、287nm附近、318nm附近及335nm附近观察到吸收峰值,在385nm附近观察到发光波长的峰值。
[0561]
实施例6
[0562]
在本实施例中,对由下述结构式(402)表示的2,5-双[3-(二苯并噻吩-4-基)苯基]嘧啶(简称:2,5mdbtp2pm-ii)的合成方法进行说明。
[0563][0564]
《2,5-双[3-(二苯并噻吩-4-基)苯基]嘧啶(简称:2,5mdbtp2pm-ii)的合成》
[0565]
(h-1)示出2,5-双[3-(二苯并噻吩-4-基)苯基]嘧啶(简称:2,5mdbtp2pm-ii)的合成方案。
[0566][0567]
在100ml茄形烧瓶中,放入0.97g(5.03mmol)的5-溴-2-氯嘧啶、3.82g(12.6mmol)3-(二苯并噻吩-4-基)苯基硼酸及2.67g(25.2mmol)碳酸钠,对该混合物加入20ml 1,3-二
甲基-3,4,5,6-四氢-2(1h)-嘧啶酮(dmpu)及10ml水。边降低压力边搅拌该混合物,来进行脱气。对该混合物加入42mg(60.3μmol)双(三苯基膦)钯(ii)二氯化物,用氩气置换空气。在氩气流下对该反应容器照射1.5小时的微波(2.45ghz,100w)来进行加热。加热后,对该混合物加入水并过滤,以得到滤渣。使用乙醇及二氯甲烷对所得到的固体进行洗涤。在该固体中加入甲苯,通过硅藻土(日本和光纯药工业株式会社、目录号码:531-16855)、矾土、硅酸镁(日本和光纯药工业株式会社、目录号码:540-00135)抽滤,浓缩滤液来得到白色固体。利用硅胶柱色谱法(作为展开剂使用甲苯)使该固体纯化。使用甲苯使得到的固体再结晶,以62%的收率得到白色固体1.87g。
[0568]
通过利用梯度升华法纯化所得到的固体1.81g。在压力为5.1pa且氩流量为10ml/min的条件下,以335℃进行加热来纯化。纯化之后,以84%的收率得到白色固体1.52g。
[0569]
通过核磁共振法(1h nmr)确认到上述化合物是目的物的2,5-双[3-(二苯并噻吩-4-基)苯基]嘧啶(简称:2,5mdbtp2pm-ii)。
[0570]
以下示出所得到的物质的1h nmr数据。
[0571]1h nmr(cdcl3,300mhz):δ= 7.46-7.51(m,4h),7.55-7.75(m,7h),7.82-7.88(m,3h),7.92(dt,j1=7.8hz,j2=1.5hz,1h),8.09(t,j1=1.5hz,1h),8.18-8.23(m,4h),8.60(dt,j1=8.4hz,j2=1.5hz,1h),8.92(t,j1=1.8hz,1h),9.17(s,2h).
[0572]
另外,图25a和图25b示出1h nmr图。另外,图25b为放大图25a的7.0ppm至9.5ppm的范围而示出的图表。
[0573]
接着,通过液相色谱-质谱联用分析(lc/ms分析)对在本实施例中得到的2,5-双[3-(二苯并噻吩-4-基)苯基]嘧啶(简称:2,5mdbtp2pm-ii)进行分析。
[0574]
在lc/ms分析中,利用沃特世(waters)公司制造的acquity uplc进行lc(液相色谱)分离,并利用沃特世公司制造的xevo g2 tof ms进行ms分析(质量分析)。lc分离中使用的色谱柱为acquity uplc beh c8(2.1
×
100mm,1.7μm),色谱柱温度为40℃。作为流动相a使用乙腈,作为流动相b使用0.1%甲酸水溶液。另外,以任意浓度将2,5mdbtp2pm-ii溶解于氯仿中并利用乙腈稀释来调节作为样品,注入量为5.0μl。
[0575]
在lc分离中利用改变流动相的组成的梯度法,检测开始后0分钟至1分钟的比为流动相a:流动相b=40:60,然后以检测开始后10分钟的比为流动相a:流动相b=95:5的方式改变组成。线性地改变组成比。
[0576]
在ms分析中利用电喷雾电离法(electrospray ionization:esi)进行离子化,毛细管电压为3.0kv,样品锥孔电压为30v,利用正模式进行检测。另外,所检测的质量范围为m/z=100至1200。
[0577]
在碰撞室(collision cell)中使在如上条件下分离并离子化了的m/z=596.14的成分碰撞到氩气来解离为产物离子。碰撞氩时的能量(碰撞能量:collision energy)为70ev。图103示出利用飞行时间质谱(tof-ms)对被解离的产物离子进行ms分析的结果。
[0578]
由图103的结果可知,在本发明的一个方式的2,5mdbtp2pm-ii中,主要在m/z=271附近、m/z=284附近检测出部分骨架的产物离子的峰值,主要在m/z=597附近检测出来源于前体离子的峰值,主要在m/z=1193附近检测出来源于二聚离子的峰值。另外,图103的结果示出来源于2,5mdbtp2pm-ii的特征,由此可以说是为了识别混合物所包含的2,5mdbtp2pm-ii而重要的数据。
[0579]
此外,图26a示出2,5mdbtp2pm-ii的甲苯溶液的吸收光谱,图26b示出其发射光谱。另外,图27a示出2,5mdbtp2pm-ii的薄膜的吸收光谱,图27b示出其发射光谱。使用紫外可见分光光度计(日本分光株式会社制造,v550型)进行吸收光谱的测量。将溶液放在石英皿中,并在石英衬底上蒸镀薄膜制造样品来进行测量。通过减去仅将甲苯放在石英皿中而测量出来的吸收光谱,获得该溶液的吸收光谱。通过减去石英衬底的吸收光谱,获得薄膜的吸收光谱。在图26a至图27b中,横轴表示波长(nm),纵轴表示强度(任意单位)。在测量甲苯溶液的情况下,在287nm附近观察到吸收峰值,在353nm附近观察到发光波长的峰值。另外,在测量薄膜的情况下,在244nm附近、268nm附近、289nm附近、326nm附近及334nm附近观察到吸收峰值,在391nm附近观察到发光波长的峰值。
[0580]
实施例7
[0581]
在本实施例中,对由下述结构式(412)表示的4,6-双[3-(2,8-二苯基-二苯并噻吩-4-基)苯基]嘧啶(简称:4,6mdbtp2pm-iii)的合成方法进行说明。
[0582][0583]
《4,6-双[3-(2,8-二苯基-二苯并噻吩-4-基)苯基]嘧啶(简称:4,6mdbtp2pm-iii)的合成》
[0584]
(i-1)示出4,6-双[3-(2,8-二苯基-二苯并噻吩-4-基)苯基]嘧啶(简称:4,6mdbtp2pm-iii)的合成方案。
[0585][0586]
在100ml圆底烧瓶中,放入0.50g(3.33mmol)的4,6-二氯嘧啶、3.80g(8.33mmol)3-(2,8-二苯基-二苯并噻吩-4-基)苯基硼酸及1.77g(16.7mmol)碳酸钠,对该混合物加入20ml1,3-二甲基-3,4,5,6-四氢-2(1h)-嘧啶酮(简称:dmpu)及10ml水。边降低压力边搅拌该混合物,来进行脱气。对该混合物加入45mg(63.7μmol)双(三苯基膦)钯(ii)二氯化物,用氩气置换空气。在氩气流下对该反应容器照射1.5小时的微波(2.45ghz,100w)来进行加热。加热后,对该混合物加入水并过滤,以得到滤渣。使用乙醇及二氯甲烷对所得到的固体进行洗涤。在该固体中加入甲苯,通过硅藻土(日本和光纯药工业株式会社、目录号码:531-16855)、矾土、硅酸镁(日本和光纯药工业株式会社、目录号码:540-00135)抽滤,浓缩滤液来得到白色固体。利用硅胶柱色谱法(作为展开剂使用甲苯)使该固体纯化。使用甲苯使得到的固体再结晶,以58%的收率得到白色固体1.74g。
[0587]
通过利用梯度升华法纯化所得到的固体1.19g。在压力为2.5pa且氩流量为5ml/min的条件下,以380℃进行加热来纯化。纯化之后,以87%的收率得到白色固体1.04g。
[0588]
通过核磁共振法(1h nmr)确认到上述化合物是目的物的4,6-双[3-(2,8-二苯基-二苯并噻吩-4-基)苯基]嘧啶(简称:4,6mdbtp2pm-iii)。
[0589]
以下示出所得到的物质的1h nmr数据。
[0590]1h nmr(cdcl3,300mhz):δ=7.37-7.42(m,4h),7.50(t,j1=7.5hz,8h),7.65-7.83(m,16h),7.97(dt,j1=7.8hz,j1=1.5hz,2h),8.29-8.33(m,3h),8.43(t,j1=1.5hz,4h),8.62(t,j1=1.5hz,2h),9.41(d,j1=1.2hz,1h).
[0591]
另外,图28a和图28b示出1h nmr图。另外,图28b为放大图28a的7.0ppm至9.5ppm的范围而示出的图表。
[0592]
接着,通过液相色谱-质谱联用分析(lc/ms分析)对在本实施例中得到的4,6-双[3-(2,8-二苯基-二苯并噻吩-4-基)苯基]嘧啶(简称:4,6mdbtp2pm-iii)进行分析。
[0593]
在lc/ms分析中,利用沃特世(waters)公司制造的acquity uplc进行lc(液相色谱)分离,并利用沃特世公司制造的xevo g2 tof ms进行ms分析(质量分析)。lc分离中使用的色谱柱为acquity uplc beh c8(2.1
×
100mm,1.7μm),色谱柱温度为40℃。作为流动相a
使用乙腈,作为流动相b使用0.1%甲酸水溶液。另外,以任意浓度将4,6mdbtp2pm-iii溶解于氯仿中并利用乙腈稀释来调节作为样品,注入量为5.0μl。
[0594]
在lc分离中利用改变流动相的组成的梯度法,检测开始后0分钟至1分钟的比为流动相a:流动相b=40:60,然后以检测开始后10分钟的比为流动相a:流动相b=95:5的方式改变组成。线性地改变组成比。
[0595]
在ms分析中利用电喷雾电离法(esi)进行离子化,毛细管电压为3.0kv,样品锥孔电压为30v,利用正模式进行检测。另外,所检测的质量范围为m/z=100至1200。
[0596]
在碰撞室(collision cell)中使在如上条件下分离并离子化了的m/z=900.26的成分碰撞到氩气来解离为产物离子。碰撞氩时的能量(碰撞能量:collision energy)为70ev。图104示出利用飞行时间质谱(tof-ms)对被解离的产物离子进行ms分析的结果。
[0597]
由图104的结果可知,在本发明的一个方式的4,6mdbtp2pm-iii中,主要在m/z=421附近、m/z=437附近及m/z=857附近检测出部分骨架的产物离子的峰值,主要在m/z=901附近检测出来源于前体离子的峰值。另外,图104的结果示出来源于4,6mdbtp2pm-iii的特征,由此可以说是为了识别混合物所包含的4,6mdbtp2pm-iii而重要的数据。
[0598]
另外,位于m/z=857附近的产物离子可以认为是因4,6mdbtp2pm-iii中的嘧啶环开环而产生的产物离子,并且这种产物离子的形式是在嘧啶环的4位及6位被取代的本发明的一个方式的有机化合物的特征之一。从而,这表示本发明的一个方式的4,6mdbtp2pm-iii包含在4位及6位被取代的嘧啶环。
[0599]
此外,图29a示出4,6mdbtp2pm-iii的甲苯溶液的吸收光谱,图29b示出其发射光谱。另外,图30a示出4,6mdbtp2pm-iii的薄膜的吸收光谱,图30b示出其发射光谱。使用紫外可见分光光度计(日本分光株式会社制造,v550型)进行吸收光谱的测量。将溶液放在石英皿中,并在石英衬底上蒸镀薄膜制造样品来进行测量。通过减去仅将甲苯放在石英皿中而测量出来的吸收光谱,获得该溶液的吸收光谱。通过减去石英衬底的吸收光谱,获得薄膜的吸收光谱。在图29a至图30b中,横轴表示波长(nm),纵轴表示强度(任意单位)。在测量甲苯溶液的情况下,在292nm附近观察到吸收峰值,在372nm附近观察到发光波长的峰值。另外,在测量薄膜的情况下,在264nm附近、301nm附近及354nm附近观察到吸收峰值,在402nm附近观察到发光波长的峰值。
[0600]
实施例8
[0601]
在本实施例中,对由下述结构式(430)表示的4,6-双[3-(二苯并呋喃-4-基)苯基]嘧啶(简称:4,6mdbfp2pm-ii)的合成方法进行说明。
[0602][0603]
《4,6-双[3-(二苯并呋喃-4-基)苯基]嘧啶(简称:4,6mdbfp2pm-ii)的合成》
[0604]
(j-1)示出4,6-双[3-(二苯并呋喃-4-基)苯基]嘧啶(简称:4,6mdbfp2pm-ii)的合
成方案。
[0605][0606]
在100ml茄形烧瓶中,放入0.79g(5.31mmol)的4,6-二氯嘧啶、3.82g(13.3mmol)3-(二苯并呋喃-4-基)-苯基硼酸及2.82g(26.6mmol)碳酸钠,对该混合物加入20ml 1,3-二甲基-3,4,5,6-四氢-2(1h)-嘧啶酮(dmpu)及10ml水。边降低压力边搅拌该混合物,来进行脱气。对该混合物加入45mg(63.7μmol)双(三苯基膦)钯(ii)二氯化物,用氩气置换空气。在氩气流下对该反应容器照射1.5小时的微波(2.45ghz,100w)来进行加热。加热后,对该混合物加入水并过滤,以得到滤渣。使用乙醇及二氯甲烷对所得到的固体进行洗涤。在该固体中加入甲苯,通过硅藻土(日本和光纯药工业株式会社、目录号码:531-16855)、矾土、硅酸镁(日本和光纯药工业株式会社、目录号码:540-00135)抽滤,浓缩滤液来得到白色固体。利用硅胶柱色谱法(作为展开剂使用甲苯)使该固体纯化。使用甲苯使得到的固体再结晶,以52%的收率得到白色固体1.56g。
[0607]
通过利用梯度升华法纯化所得到的固体1.51g。在压力为3.5pa且氩流量为5ml/min的条件下,以280℃进行加热来纯化。纯化之后,以81%的收率得到白色固体1.23g。
[0608]
通过核磁共振法(1h nmr)确认到上述化合物是目的物的4,6-双[3-(二苯并呋喃-4-基)苯基]嘧啶(简称:4,6mdbfp2pm-ii)。
[0609]
以下示出所得到的物质的1h nmr数据。
[0610]1h nmr(cdcl3,300mhz):δ=7.34-7.50(m,6h),7.57(d,j1=8.4hz,2h),7.73(t,j1=7.8hz,4h),7.98-8.01(m,4h),8.11(d,j1=7.8hz,2h),8.26(d,j1=7.8hz,2h),8.33(d,j1=0.9hz,1h),8.69(t,j1=1.5hz,2h),9.41(s,1h).
[0611]
另外,图31a和图31b示出1h nmr图。另外,图31b为放大图31a的7.0ppm至9.5ppm的范围而示出的图表。
[0612]
接着,通过液相色谱-质谱联用分析(lc/ms分析)对在本实施例中得到的4,6-双[3-(二苯并呋喃-4-基)苯基]嘧啶(简称:4,6mdbfp2pm-ii)进行分析。
[0613]
在lc/ms分析中,利用沃特世(waters)公司制造的acquity uplc进行lc(液相色谱)分离,并利用沃特世公司制造的xevo g2 tof ms进行ms分析(质量分析)。lc分离中使用的色谱柱为acquity uplc beh c8(2.1
×
100mm,1.7μm),色谱柱温度为40℃。作为流动相a使用乙腈,作为流动相b使用0.1%甲酸水溶液。另外,以任意浓度将4,6mdbfp2pm-ii溶解于氯仿中并利用乙腈稀释来调节作为样品,注入量为5.0μl。
[0614]
在lc分离中利用改变流动相的组成的梯度法,检测开始后0分钟至1分钟的比为流动相a:流动相b=40:60,然后以检测开始后10分钟的比为流动相a:流动相b=95:5的方式改变组成。线性地改变组成比。
[0615]
在ms分析中利用电喷雾电离法(esi)进行离子化,毛细管电压为3.0kv,样品锥孔电压为30v,利用正模式进行检测。另外,所检测的质量范围为m/z=100至1200。
[0616]
在碰撞室(collision cell)中使在如上条件下分离并离子化了的m/z=564.18的成分碰撞到氩气来解离为产物离子。碰撞氩时的能量(碰撞能量:collision energy)为70ev。图105示出利用飞行时间质谱(tof-ms)对被解离的产物离子进行ms分析的结果。
[0617]
由图105的结果可知,在本发明的一个方式的4,6mdbfp2pm-ii中,主要在m/z=215附近、m/z=239附近、m/z=270附近及m/z=521附近检测出部分骨架的产物离子的峰值,主要在m/z=565附近检测出来源于前体离子的峰值,主要在m/z=1129附近检测出来源于二聚离子的峰值。另外,图105的结果示出来源于4,6mdbfp2pm-ii的特征,由此可以说是为了识别混合物所包含的4,6mdbfp2pm-ii而重要的数据。
[0618]
另外,位于m/z=521附近的产物离子可以认为是因4,6mdbfp2pm-ii中的嘧啶环开环而产生的产物离子,并且这种产物离子的形式是在嘧啶环的4位及6位被取代的本发明的一个方式的有机化合物的特征之一。从而,这表示本发明的一个方式的4,6mdbfp2pm-ii包含在4位及6位被取代的嘧啶环。
[0619]
此外,图32a示出4,6mdbfp2pm-ii的甲苯溶液的吸收光谱,图32b示出其发射光谱。另外,图33a示出4,6mdbfp2pm-ii的薄膜的吸收光谱,图33b示出其发射光谱。使用紫外可见分光光度计(日本分光株式会社制造,v550型)进行吸收光谱的测量。将溶液放在石英皿中,并在石英衬底上蒸镀薄膜制造样品来进行测量。通过减去仅将甲苯放在石英皿中而测量出来的吸收光谱,获得该溶液的吸收光谱。通过减去石英衬底的吸收光谱,获得薄膜的吸收光谱。在图32a至图33b中,横轴表示波长(nm),纵轴表示强度(任意单位)。在测量甲苯溶液的情况下,在289nm附近观察到吸收峰值,在383nm附近观察到发光波长的峰值。另外,在测量薄膜的情况下,在206nm附近、248nm附近、290nm附近、303nm附近及315nm附近观察到吸收峰值,在383nm附近观察到发光波长的峰值。
[0620]
实施例9
[0621]
在本实施例中,对由下述结构式(431)表示的2,4-双[3-(二苯并呋喃-4-基)苯基]嘧啶(简称:2,4mdbfp2pm-ii)的合成方法进行说明。
[0622][0623]
《2,4-双[3-(二苯并呋喃-4-基)苯基]嘧啶(简称:2,4mdbfp2pm-ii)的合成》
[0624]
(k-1)示出2,4-双[3-(二苯并呋喃-4-基)苯基]嘧啶(简称:2,4mdbfp2pm-ii)的合成方案。
[0625][0626]
在100ml茄形烧瓶中,放入0.79g(5.31mmol)的2,4-二氯嘧啶、3.82g(13.3mmol)3-(二苯并呋喃-4-基)-苯基硼酸及2.82g(26.6mmol)碳酸钠,对该混合物加入20ml1,3-二甲基-3,4,5,6-四氢-2(1h)-嘧啶酮(dmpu)及10ml水。边降低压力边搅拌该混合物,来进行脱气。对该混合物加入45mg(63.7μmol)双(三苯基膦)钯(ii)二氯化物,用氩气置换空气。在氩气流下对该反应容器照射1小时的微波(2.45ghz,100w)来进行加热。加热后,对该混合物加入水并过滤,以得到滤渣。使用乙醇及二氯甲烷对所得到的固体进行洗涤。在该固体中加入甲苯,通过硅藻土(日本和光纯药工业株式会社、目录号码:531-16855)、矾土、硅酸镁(日本和光纯药工业株式会社、目录号码:540-00135)抽滤,浓缩滤液来得到白色固体。利用硅胶柱色谱法(作为展开剂使用甲苯)使该固体纯化。使用甲苯使得到的固体再结晶,以63%的收率得到白色固体1.89g。
[0627]
通过利用梯度升华法纯化所得到的固体1.93g。在压力为3.3pa且氩流量为5ml/min的条件下,以270℃进行加热来纯化。纯化之后,以74%的收率得到白色固体1.43g。
[0628]
通过核磁共振法(1h nmr)确认到上述化合物是目的物的2,4-双[3-(二苯并呋喃-4-基)苯基]嘧啶(简称:2,4mdbfp2pm-ii)。
[0629]
以下示出所得到的物质的1h nmr数据。
[0630]1h nmr(cdcl3,300mhz):δ=7.31-7.45(m,6h),7.54-7.57(m,2h),7.68-7.77(m,5h),7.92-8.00(m,4h),8.07-8.13(m,2h),8.33(dt,j1=7.8hz,j2=1.2hz,1h),8.70(dt,j1=8.1hz,j2=1.5hz,1h),8.89(t,j1=1.5hz,1h),8.93(d,j1=5.4hz,1h),9.22(t,j1=1.5hz,1h).
[0631]
另外,图34a和图34b示出1h nmr图。另外,图34b为放大图34a的7.0ppm至9.5ppm的范围而示出的图表。
[0632]
接着,通过液相色谱-质谱联用分析(lc/ms分析)对在本实施例中得到的2,4-双[3-(二苯并呋喃-4-基)苯基]嘧啶(简称:2,4mdbfp2pm-ii)进行分析。
[0633]
在lc/ms分析中,利用沃特世(waters)公司制造的acquity uplc进行lc(液相色谱)分离,并利用沃特世公司制造的xevo g2 tof ms进行ms分析(质量分析)。lc分离中使用
的色谱柱为acquity uplc beh c8(2.1
×
100mm,1.7μm),色谱柱温度为40℃。作为流动相a使用乙腈,作为流动相b使用0.1%甲酸水溶液。另外,以任意浓度将2,4mdbfp2pm-ii溶解于氯仿中并利用乙腈稀释来调节作为样品,注入量为5.0μl。
[0634]
在lc分离中利用改变流动相的组成的梯度法,检测开始后0分钟至1分钟的比为流动相a:流动相b=40:60,然后以检测开始后10分钟的比为流动相a:流动相b=95:5的方式改变组成。线性地改变组成比。
[0635]
在ms分析中利用电喷雾电离法(esi)进行离子化,毛细管电压为3.0kv,样品锥孔电压为30v,利用正模式进行检测。另外,所检测的质量范围为m/z=100至1200。
[0636]
在碰撞室(collision cell)中使在如上条件下分离并离子化了的m/z=564.18的成分碰撞到氩气来解离为产物离子。碰撞氩时的能量(碰撞能量:collision energy)为70ev。图106示出利用飞行时间质谱(tof-ms)对被解离的产物离子进行ms分析的结果。
[0637]
由图106的结果可知,在本发明的一个方式的2,4mdbfp2pm-ii中,主要在m/z=239附近、m/z=268附近检测出部分骨架的产物离子的峰值,主要在m/z=565附近检测出来源于前体离子的峰值,主要在m/z=1129附近检测出来源于二聚离子的峰值。另外,图106的结果示出来源于2,4mdbfp2pm-ii的特征,由此可以说是为了识别混合物所包含的2,4mdbfp2pm-ii而重要的数据。
[0638]
此外,图35a示出2,4mdbfp2pm-ii的甲苯溶液的吸收光谱,图35b示出其发射光谱。另外,图36a示出2,4mdbfp2pm-ii的薄膜的吸收光谱,图36b示出其发射光谱。使用紫外可见分光光度计(日本分光株式会社制造,v550型)进行吸收光谱的测量。将溶液放在石英皿中,并在石英衬底上蒸镀薄膜制造样品来进行测量。通过减去仅将甲苯放在石英皿中而测量出来的吸收光谱,获得该溶液的吸收光谱。通过减去石英衬底的吸收光谱,获得薄膜的吸收光谱。在图35a至图36b中,横轴表示波长(nm),纵轴表示强度(任意单位)。在测量甲苯溶液的情况下,在283nm附近观察到吸收峰值,在413nm附近观察到发光波长的峰值。另外,在测量薄膜的情况下,在316nm附近观察到吸收峰值,在387nm附近观察到发光波长的峰值。
[0639]
实施例10
[0640]
在本实施例中,对由下述结构式(432)表示的2,5-[3-(二苯并呋喃-4-基)苯基]嘧啶(简称:2,5mdbfp2pm-ii)的合成方法进行说明。
[0641][0642]
《2,5-[3-(二苯并呋喃-4-基)苯基]嘧啶(简称:2,5mdbfp2pm-ii)的合成》
[0643]
(l-1)示出2,5-[3-(二苯并呋喃-4-基)苯基]嘧啶(简称:2,5mdbfp2pm-ii)的合成方案。
[0644][0645]
在100ml茄形烧瓶中,放入1.03g(5.31mmol)的5-溴-2-氯嘧啶、3.82g(13.3mmol)3-(二苯并呋喃-4-基)-苯基硼酸及2.82g(26.6mmol)碳酸钠,对该混合物加入20ml1,3-二甲基-3,4,5,6-四氢-2(1h)-嘧啶酮(dmpu)及10ml水。边降低压力边搅拌该混合物,来进行脱气。对该混合物加入45mg(63.7μmol)双(三苯基膦)钯(ii)二氯化物,用氩气置换空气。在氩气流下对该反应容器照射2.5小时的微波(2.45ghz,100w)来进行加热。加热后,对该混合物加入水并过滤,以得到滤渣。使用乙醇及二氯甲烷对所得到的固体进行洗涤。在该固体中加入甲苯,通过硅藻土(日本和光纯药工业株式会社、目录号码:531-16855)、矾土、硅酸镁(日本和光纯药工业株式会社、目录号码:540-00135)抽滤,浓缩滤液来得到白色固体。利用硅胶柱色谱法(作为展开剂使用甲苯)使该固体纯化。使用甲苯使得到的固体再结晶,以60%的收率得到白色固体1.80g。
[0646]
通过利用梯度升华法纯化所得到的固体1.80g。在压力为3.3pa且氩流量为5ml/min的条件下,以300℃进行加热来纯化。纯化之后,以84%的收率得到白色固体1.51g。
[0647]
通过核磁共振法(1h nmr)确认到上述化合物是目的物的2,5-[3-(二苯并呋喃-4-基)苯基]嘧啶(简称:2,5mdbfp2pm-ii)。
[0648]
以下示出所得到的物质的1h nmr数据。
[0649]1h nmr(cdcl3,300mhz):δ=7.35-7.41(m,2h),7.45-7.52(m,4h),7.62-7.78(m,7h),7.96-8.03(m,5h),8.14(dt,j1=7.8hz,j2=1.5hz,1h),8.21(dd,j1=2.7hz,j2=1.5hz,1h),8.59(dt,j1=7.8hz,j2=1.5hz,1h),9.03(t,j1=1.2hz,1h),9.18(s,2h).
[0650]
另外,图37a和图37b示出1h nmr图。另外,图37b为放大图37a的7.0ppm至9.5ppm的范围而示出的图表。
[0651]
接着,通过液相色谱-质谱联用分析(lc/ms分析)对在本实施例中得到的2,5-[3-(二苯并呋喃-4-基)苯基]嘧啶(简称:2,5mdbfp2pm-ii)进行分析。
[0652]
在lc/ms分析中,利用沃特世(waters)公司制造的acquity uplc进行lc(液相色谱)分离,并利用沃特世公司制造的xevo g2 tof ms进行ms分析(质量分析)。lc分离中使用的色谱柱为acquity uplc beh c8(2.1
×
100mm,1.7μm),色谱柱温度为40℃。作为流动相a使用乙腈,作为流动相b使用0.1%甲酸水溶液。另外,以任意浓度将2,5mdbfp2pm-ii溶解于
氯仿中并利用乙腈稀释来调节作为样品,注入量为5.0μl。
[0653]
在lc分离中利用改变流动相的组成的梯度法,检测开始后0分钟至1分钟的比为流动相a:流动相b=40:60,然后以检测开始后10分钟的比为流动相a:流动相b=95:5的方式改变组成。线性地改变组成比。
[0654]
在ms分析中利用电喷雾电离法(esi)进行离子化,毛细管电压为3.0kv,样品锥孔电压为30v,利用正模式进行检测。另外,所检测的质量范围为m/z=100至1200。
[0655]
在碰撞室(collision cell)中使在如上条件下分离并离子化了的m/z=564.18的成分碰撞到氩气来解离为产物离子。碰撞氩时的能量(碰撞能量:collision energy)为70ev。图107示出利用飞行时间质谱(tof-ms)对被解离的产物离子进行ms分析的结果。
[0656]
由图107的结果可知,在本发明的一个方式的2,5mdbfp2pm-ii中,主要在m/z=239附近、m/z=255附近及m/z=268附近检测出部分骨架的产物离子的峰值,主要在m/z=565附近检测出来源于前体离子的峰值,主要在m/z=1129附近检测出来源于二聚离子的峰值。另外,图107的结果示出来源于2,5mdbfp2pm-ii的特征,由此可以说是为了识别混合物所包含的2,5mdbfp2pm-ii而重要的数据。
[0657]
此外,图38a示出2,5mdbfp2pm-ii的甲苯溶液的吸收光谱,图38b示出其发射光谱。另外,图39a示出2,5mdbfp2pm-ii的薄膜的吸收光谱,图39b示出其发射光谱。使用紫外可见分光光度计(日本分光株式会社制造,v550型)进行吸收光谱的测量。将溶液放在石英皿中,并在石英衬底上蒸镀薄膜制造样品来进行测量。通过减去仅将甲苯放在石英皿中而测量出来的吸收光谱,获得该溶液的吸收光谱。通过减去石英衬底的吸收光谱,获得薄膜的吸收光谱。在图38a至图39b中,横轴表示波长(nm),纵轴表示强度(任意单位)。在测量甲苯溶液的情况下,在287nm附近观察到吸收峰值,在425nm附近观察到发光波长的峰值。另外,在测量薄膜的情况下,在205nm附近、247nm附近、280nm附近、302nm附近及316nm附近观察到吸收峰值,在390nm附近观察到发光波长的峰值。
[0658]
如实施例1至实施例10示出,根据吸收光谱,本发明的第二化合物的包含嘧啶骨架的有机化合物的homo与lumo之间的带隙(bg)宽。将该有机化合物在可见区域中几乎不具有吸收并具有高透光性,所以当将该有机化合物用于发光元件时,不容易吸收发光能量,从而可以期待获得高效率的元件。并且,根据吸收光谱,该有机化合物具有高s1能级,所以可以期待将该有机化合物用作发射可见区域的荧光的材料的主体材料。另外,因为该有机化合物具有高s1能级,所以可以期待具有高t1能级。此外,因为该有机化合物发射紫色至蓝色的光,所以也可以将其用作发光材料。
[0659]
实施例11
[0660]
在本实施例中,对如下化合物进行电化学特性(溶液)测定:作为第二化合物的包含嘧啶骨架的有机化合物的一个方式,即在实施例1至10中制造的由结构式(300)、(311)、(314)、(400)、(401)、(402)、(412)、(430)、(431)及(432)表示的化合物;可用作第一化合物的由结构式(200)至(204)、(210)至(213)、(250)及(251)表示的化合物。下面示出在本实施例中使用的材料的化学式。
[0661]
[0662]
[0663]
[0664][0665]
另外,作为测定方法,采用循环伏安(cv)测定。电化学分析仪(bas株式会社(bas inc.)制造,als型号600a或600c)用于该测定。表1示出测定结果。
[0666]
[0667][0668]
nd:no data
[0669]
表1所示的氧化一侧(单电子氧化的电位)的数据相当于homo能级的值。由于第二化合物的包含嘧啶骨架的有机化合物的一个方式的4,6mpnp2pm等的homo能级深于作为掺杂物的铱金属配合物的homo能级(4,6mpnp2pm等的homo能级的值小于铱金属配合物的homo能级的值),因此能够将空穴有效地注入到作为掺杂物的这些铱金属配合物。另外,当铱金属配合物的homo能级为-6.0ev至-5.0ev时,与第二化合物的包含嘧啶骨架的有机化合物相比,该铱金属配合物更有效地捕获空穴。
[0670]
另外,表1所示的还原一侧(单电子还原的电位)的数据相当于lumo能级的值。由于第二化合物的包含嘧啶骨架的有机化合物的一个方式的4,6mpnp2pm等的lumo能级等于或较浅于作为掺杂物的磷光铱金属配合物的lumo能级,这些数值相近,因此能够将电子有效地注入到作为掺杂物的铱金属配合物。另外,由此可以说能够制造驱动电压低的元件,而作为掺杂物的铱配合物不妨碍主体材料的4,6mpnp2pm等的电子传输性。
[0671]
根据上述结果,本发明的包含嘧啶骨架的有机化合物具有深homo能级及适当的lumo能级,所以适合用作铱金属配合物的主体材料。尤其适合用作具有与主体材料相近的lumo能级的二嗪骨架的铱金属配合物,具体地说具有嘧啶骨架的铱金属配合物。
[0672]
另外,由结构式(200)至(204)、(250)及(251)表示的磷光铱金属配合物具有嘧啶骨架,由结构式(210)至(213)表示的磷光铱金属配合物具有吡嗪骨架,所以上述化合物都具有二嗪骨架。
[0673]
另外,上述循环伏安法(cv)的测定方法如下。虽然在下面说明中提到4,6mpnp2pm,
但是利用同样方法对其他化合物进行测定。
[0674]
对用于cv测定的溶液,脱水的二甲基甲酰胺(dmf,西格玛-奥尔德里奇公司(sigma-aldrich inc.)制造,99.8%,目录号码22705-6)用作溶剂,高氯酸四正丁基铵(n-bu4nclo4,东京化学工业公司,目录号码t0836)是支持电解质(supporting electrolyte),溶解于溶剂使高氯酸四正丁基铵的浓度为100mmol/l。此外,将需测量的目标物溶解于溶剂中,使其浓度为2mmol/l。另外,作为工作电极使用铂电极(bas株式会社(bas inc.)制造,pte铂电极),作为辅助电极使用铂电极(bas株式会社(bas inc.)制造,vc-3用pt对电极(5cm)),作为参考电极使用ag/ag 电极(bas株式会社(bas inc.)制造,re7非水溶剂型参考电极)。另外,在室温下(20℃至25℃)进行cv测定。另外,将cv测定时的扫描速度统一为0.1v/sec。
[0675]
(参考电极相对于真空能级的势能的计算)
[0676]
首先,算出在本实施例中使用的参考电极(ag/ag 电极)的相对于真空能级的势能(ev)。即,算出ag/ag 电极的费米能级。已知相对于标准氢电极,甲醇中的二茂铁的氧化还原电势是 0.610[v vs.she](参考文献:christian r.goldsmith et al.,j.am.chem.soc.,vol.124,no.1,pp.83-96,2002)。
[0677]
另一方面,通过利用在本实施例中使用的参考电极来算出在甲醇中的二茂铁的氧化还原电位,其结果是 0.11v[vs.ag/ag ]。因此,发现在本实施例中使用的参考电极的势能比标准氢电极低0.50[ev]。
[0678]
这里,已知标准氢电极的相对于真空能级的势能是-4.44ev(参考文献:toshihiro ohnishi,tamami koyama.high molecular el material.,kyoritsu shuppan,pp.64-67)。因此,算出在本实施例中使用的参考电极相对于真空能级的势能为-4.44-0.50=-4.94[ev]。
[0679]
在本实施例的化合物的氧化反应特性的测定中,在将工作电极对参考电极的电位从0.3v左右扫描到1.5v左右之后从1.5v左右扫描到0.3v左右。
[0680]
接着,对根据目的物的cv测定的homo能级的算出进行详细的说明。算出氧化峰电位(从中性态至氧化侧之间)e
pa
[v]和还原峰电位(从氧化侧至中性态之间)e
pc
[v]。从而可以算出半波电位(位于e
pa
与e
pc
的中间的电位)为(e
pa
e
pc
)/2[v]。这示出实施例的化合物由半波电位的值[v vs.ag/ag ]的电能氧化,该能量相当于homo能级。
[0681]
在本实施例的化合物的还原反应特性的测定中,在将工作电极对参考电极的电位从-1.5v左右扫描到-2.2v左右之后从-2.2v左右扫描到-1.5v左右。
[0682]
接着,对根据目的物的cv测定的lumo能级的算出进行详细的说明。算出还原峰电位(从中性态至还原侧之间)e
pa
[v]和氧化峰电位(从还原侧至中性态之间)e
pc
[v]。从而可以算出半波电位(位于e
pa
与e
pc
的中间的电位)为(e
pa
e
pc
)/2[v]。这示出实施例的化合物由半波电位的值[v vs.ag/ag ]的电能还原,该能量相当于lumo能级。
[0683]
实施例12
[0684]
在本实施例中,评价使用第一化合物及第二化合物作为发光物质的发光元件1,该第一化合物为包含嘧啶骨架的磷光铱金属配合物,而该第二化合物为实施方式1及实施例1所示的由结构式(300)表示的包含嘧啶骨架的有机化合物。以下,示出在本实施例中使用的材料的化学式。
[0685][0686]
参照图40说明发光元件1。以下,示出本实施例的发光元件1的制造方法。
[0687]
(发光元件1)
[0688]
首先,在衬底1100上通过溅射法形成包含硅或氧化硅的氧化铟-氧化锡化合物(ito-sio2,以下简称为itso),由此形成第一电极1101。另外,所使用的靶材的组成比为in2o3:sno2:sio2=85:10:5[wt.%]。另外,将第一电极1101的厚度设定为110nm,且将其电极面积设定为2mm
×
2mm。这里,第一电极1101是用作发光元件的阳极的电极。
[0689]
接着,作为用来在衬底1100上形成发光元件的预处理,在用水洗涤衬底表面并在200℃下进行焙烧1小时之后,进行uv臭氧处理370秒。
[0690]
然后,将衬底放进到真空蒸镀装置中,其内部被减压到10-4
pa左右。在真空蒸镀装置内的加热室中,在170℃下进行30分钟的真空焙烧,然后使衬底1100冷却30分钟左右。
[0691]
接着,以使形成有第一电极1101的面朝下的方式将形成有第一电极1101的衬底1100固定在设置在真空蒸镀装置内的衬底支架上,并将压力降低到10-4
pa左右,然后在第一
电极1101上共蒸镀1,3,5-三(二苯并噻吩-4-基)-苯(简称:dbt3p-ii)和氧化钼,从而形成空穴注入层1111。将空穴注入层1111的厚度设定为40nm,将dbt3p-ii与氧化钼的重量比调节为4:2(=dbt3p-ii:氧化钼)。另外,共蒸镀法是指在一个处理室中从多个蒸发源同时进行蒸镀的蒸镀法。
[0692]
接着,在空穴注入层1111上形成厚度为20nm的4-苯基-4'-(9-苯基芴-9-基)三苯胺(简称:bpaflp),由此形成空穴传输层1112。
[0693]
再者,通过共蒸镀在实施例1中合成的4,6-双[3-(菲-9-基)苯基]嘧啶(简称:4,6mpnp2pm)、4-苯基-4
′‑
(9-苯基-9h-咔唑-3-基)三苯胺(简称:pcba1bp)以及(乙酰丙酮合)双(6-叔丁基-4-苯基嘧啶合)铱(iii)(简称:[ir(tbuppm)2(acac)]),在空穴传输层1112上形成第一发光层1113a。这里,将4,6mpnp2pm、pcba1bp以及[ir(tbuppm)2(acac)]的重量比调节为0.7:0.3:0.05(=4,6mpnp2pm:pcba1bp:[ir(tbuppm)2(acac)])。另外,将第一发光层1113a的厚度设定为15nm。
[0694]
接着,通过在第一发光层1113a上共蒸镀4,6mpnp2pm、pcba1bp以及[ir(tbuppm)2(acac)],在第一发光层1113a上形成第二发光层1113b。这里,将4,6mpnp2pm、pcba1bp以及[ir(tbuppm)2(acac)]的重量比调节为0.8:0.2:0.05(=4,6mpnp2pm:pcba1bp:[ir(tbuppm)2(acac)])。另外,将第二发光层1113b的厚度设定为25nm。
[0695]
接着,在第二发光层1113b上形成厚度为10nm的4,6mpnp2pm,由此形成第一电子传输层1114a。
[0696]
接着,在第一电子传输层1114a上形成厚度为20nm的形成红菲绕啉(简称:bphen),由此形成第二电子传输层1114b。
[0697]
再者,在第二电子传输层1114b上蒸镀形成厚度为1nm的氟化锂(lif)膜,由此形成电子注入层1115。
[0698]
最后,作为用作阴极的第二电极1103,通过蒸镀形成厚度为200nm的铝膜,由此制造本实施例的发光元件1。
[0699]
表2示出经上述步骤而得到的发光元件1的元件结构。
[0700][0701]
在氮气氛的手套箱中,以不使发光元件1暴露于大气的方式对发光元件1进行密封处理(将密封材料涂敷在元件的周围,在密封时以80℃进行1小时的热处理)。然后,测定该
发光元件1的工作特性。另外,在室温下(在保持为25℃的气氛中)进行测定。
[0702]
图41示出发光元件1的电流密度-亮度特性。在图41中,横轴表示电流密度(ma/cm2),而纵轴表示亮度(cd/m2)。另外,图42示出发光元件1的电压-亮度特性。在图42中,横轴表示电压(v),而纵轴表示亮度(cd/m2)。另外,图43示出发光元件1的亮度-电流效率特性。在图43中,横轴表示亮度(cd/m2),而纵轴表示电流效率(cd/a)。另外,图44示出发光元件1的电压-电流特性。在图44中,横轴表示电压(v),而纵轴表示电流(ma)。另外,图45示出发光元件1的亮度-色度坐标特性。在图45中,横轴表示亮度(cd/m2),而纵轴表示色度(x坐标及y坐标)。另外,图46示出发光元件1的亮度-功率效率特性。在图46中,横轴表示亮度(cd/m2),而纵轴表示功率效率(lm/w)。
[0703]
另外,表3示出发光元件1的亮度大约为1000cd/m2时的电压(v)、电流密度(ma/cm2)、cie色度坐标(x,y)、亮度(cd/m2)、电流效率(cd/a)和外部量子效率(%)。
[0704]
[表3]
[0705][0706]
图47示出发光元件1的电流密度为2.5ma/cm2时的发射光谱。如图47所示,发光元件1的发射光谱在547nm处具有峰值。
[0707]
另外,如表3所示,发光元件1的亮度为1300cd/m2时的cie色度坐标为(x,y)=(0.43,0.56)。由此可知,得到来源于掺杂剂的发光。
[0708]
根据上述可知,将作为第一化合物的包含嘧啶骨架的磷光铱金属配合物及作为第二化合物的包含嘧啶骨架的有机化合物用作发光物质的本发明的一个方式的发光元件1能够高效地发射绿色波长区的光。另外,4,6mpnp2pm具有高于绿色光的t1能级,因此,4,6mpnp2pm优选用作发射绿色光或比绿色光更长的波长的光的发光材料的主体材料。
[0709]
另外,根据图41及图42可知,发光元件1是低驱动电压及低耗电量的元件。另外,根据图43及图46可知,发光元件1是高效率元件。根据图45可知,发光元件1是各亮度上的载流子平衡优良的元件。
[0710]
接着,对上述发光元件1进行可靠性试验的评价。图48及图49示出可靠度试验的结果。
[0711]
在可靠性试验中,在将初始亮度设定为5000cd/m2并电流密度恒定的条件下驱动发光元件1。图48示出其结果。横轴表示元件的驱动时间(h),而纵轴表示起始亮度为100%时的归一化亮度(%)。根据图48可知,大约290小时后,发光元件1的归一化亮度降低到70%以下。
[0712]
根据图48可知,发光元件1是长使用寿命的元件。
[0713]
根据上述结果可知,将作为第一化合物的包含嘧啶的磷光铱金属配合物及作为第二化合物的包含嘧啶骨架的有机化合物用作发光物质的本发明的一个方式的发光元件1是
高效率、低驱动电压、低耗电量以及长使用寿命的发光元件。
[0714]
接着,在可靠性试验中,在将初始亮度设定为5000cd/m2并电流密度恒定的条件下测定发光元件1的电压的随时间变化。图49示出其结果。横轴表示元件的驱动时间(h),而纵轴表示电压(v)。根据图49可知,发光元件1的电压的随时间上升的程度小。
[0715]
实施例13
[0716]
在本实施例中,评价使用本发明的一个方式的在实施例4中合成的由结构式(400)表示的有机化合物的发光元件2及发光元件3。以下,示出在本实施例中使用的发光元件2的材料的化学式。
[0717][0718]
参照图50a说明发光元件2。以下,示出本实施例的发光元件2的制造方法。
[0719]
(发光元件2)
[0720]
首先,在衬底2100上通过溅射法形成包含硅或氧化硅的氧化铟-氧化锡化合物(itso),由此形成第一电极2101。另外,所使用的靶材的组成比为in2o3:sno2:sio2=85:10:
5[wt.%]。另外,将第一电极2101的厚度设定为110nm,且将其电极面积设定为2mm
×
2mm。这里,第一电极2101是用作发光元件的阳极的电极。
[0721]
接着,作为用来在衬底2100上形成发光元件的预处理,在用水洗涤衬底表面并在200℃下进行焙烧1小时之后,进行uv臭氧处理370秒。
[0722]
然后,将衬底放进到真空蒸镀装置中,其内部被减压到10-4
pa左右。在真空蒸镀装置内的加热室中,在170℃下进行30分钟的真空焙烧,然后使衬底2100冷却30分钟左右。
[0723]
接着,以使形成有第一电极2101的面朝下的方式将形成有第一电极2101的衬底2100固定在设置在真空蒸镀装置内的衬底支架上,并将压力降低到10-4
pa左右,然后在第一电极2101上共蒸镀1,3,5-三(二苯并噻吩-4-基)-苯(简称:dbt3p-ii)和氧化钼,从而形成空穴注入层2111。将空穴注入层2111的厚度设定为40nm,将dbt3p-ii与氧化钼的重量比调节为4:2(=dbt3p-ii:氧化钼)。另外,共蒸镀法是指在一个处理室中从多个蒸发源同时进行蒸镀的蒸镀法。
[0724]
接着,在空穴注入层2111上形成厚度为20nm的4-苯基-4'-(9-苯基芴-9-基)三苯胺(简称:bpaflp),由此形成空穴传输层2112。
[0725]
再者,通过共蒸镀在实施例4中合成的4,6-双[3-(二苯并噻吩-4-基)苯基]嘧啶(简称:4,6mdbtp2pm-ii)、4-苯基-4
′‑
(9-苯基-9h-咔唑-3-基)三苯胺(简称:pcba1bp)以及(乙酰丙酮合)双(6-叔丁基-4-苯基嘧啶合)铱(iii)(简称:[ir(tbuppm)2(acac)]),在空穴传输层2112上形成第一发光层2113a。这里,将4,6mdbtp2pm-ii、pcba1bp以及[ir(tbuppm)2(acac)]的重量比调节为0.7:0.3:0.05(=4,6mdbtp2pm-ii:pcba1bp:[ir(tbuppm)2(acac)])。另外,将第一发光层2113a的厚度设定为15nm。
[0726]
接着,通过在第一发光层2113a上共蒸镀4,6mdbtp2pm-ii、pcba1bp以及[ir(tbuppm)2(acac)],在第一发光层2113a上形成第二发光层2113b。这里,将4,6mdbtp2pm-ii、pcba1bp以及[ir(tbuppm)2(acac)]的重量比调节为0.8:0.2:0.05(=4,6mdbtp2pm-ii:pcba1bp:[ir(tbuppm)2(acac)])。另外,将第二发光层2113b的厚度设定为25nm。
[0727]
接着,在第二发光层2113b上形成厚度为10nm的4,6mdbtp2pm-ii,由此形成第一电子传输层2114a。
[0728]
接着,在第一电子传输层2114a上形成厚度为20nm的红菲绕啉(简称:bphen),由此形成第二电子传输层2114b。
[0729]
再者,在第二电子传输层2114b上蒸镀形成厚度为1nm的氟化锂(lif),由此形成电子注入层2115。
[0730]
最后,作为用作阴极的第二电极2103,通过蒸镀形成厚度为200nm的铝膜,由此制造本实施例的发光元件2。
[0731]
表4示出经上述步骤而得到的发光元件2的元件结构。
[0732][0733]
以下,示出在本实施例中使用的发光元件3的材料的化学式。
[0734][0735]
参照图50b说明发光元件3。以下,示出本实施例的发光元件3的制造方法。
[0736]
(发光元件3)
[0737]
首先,在衬底2100上通过溅射法形成包含硅或氧化硅的氧化铟-氧化锡化合物(itso),由此形成第一电极2101。另外,所使用的靶材的组成比为in2o3:sno2:sio2=85:10:5[wt.%]。另外,将第一电极2101的厚度设定为110nm,且将其电极面积设定为2mm
×
2mm。这里,第一电极2101是用作发光元件的阳极的电极。
[0738]
接着,作为用来在衬底2100上形成发光元件的预处理,在用水洗涤衬底表面并在200℃下进行焙烧1小时之后,进行uv臭氧处理370秒。
[0739]
然后,将衬底放进到真空蒸镀装置中,其内部被减压到10-4
pa左右。在真空蒸镀装置内的加热室中,在170℃下进行30分钟的真空焙烧,然后使衬底2100冷却30分钟左右。
[0740]
接着,以使形成有第一电极2101的面朝下的方式将形成有第一电极2101的衬底2100固定在设置在真空蒸镀装置内的衬底支架上,并将压力降低到10-4
pa左右,然后在第一
电极2101上共蒸镀4,4
′‑
二(n-咔唑基)联苯(简称:cbp)和氧化钼,从而形成空穴注入层2111。将空穴注入层2111的厚度设定为60nm,将cbp与氧化钼的重量比调节为4:2(=cbp:氧化钼)。另外,共蒸镀法是指在一个处理室中从多个蒸发源同时进行蒸镀的蒸镀法。
[0741]
接着,在空穴注入层2111上形成厚度为20nm的1,3-双(n-咔唑基)苯(简称:mcp),由此形成空穴传输层2112。
[0742]
再者,通过共蒸镀在实施例4中合成的4,6-双[3-(二苯并噻吩-4-基)苯基]嘧啶(简称:4,6mdbtp2pm-ii)、9-苯基-9h-3-(9-苯基-9h-咔唑-3-基)咔唑(简称:pccp)以及三[3-甲基-1-(2-甲基苯基)-5-苯基-1h-1,2,4-三唑合(triazolato)]铱(iii)(简称:[ir(mptz1-mp)3]),在空穴传输层2112上形成第一发光层2113a。这里,将4,6mdbtp2pm-ii、pccp以及[ir(mptz1-mp)3]的重量比调节为1:0.3:0.08(=4,6mdbtp2pm-ii:pccp:[ir(mptz1-mp)3])。另外,将第一发光层2113a的厚度设定为30nm。
[0743]
接着,通过在第一发光层2113a上共蒸镀2-[3-(二苯并噻吩-4-基)苯基]-1-苯基-1h-苯并咪唑(简称:mdbtbim-ii)以及[ir(mptz1-mp)3],在第一发光层2113a上形成第二发光层2113b。这里,将mdbtbim-ii以及[ir(mptz1-mp)3]的重量比调节为1:0.08(=mdbtbim-ii:[ir(mptz1-mp)3])。另外,将第二发光层2113b的厚度设定为10nm。
[0744]
接着,在第二发光层2113b上形成厚度为15nm的红菲绕啉(简称:bphen),由此形成电子传输层2114。
[0745]
再者,在电子传输层2114上蒸镀形成厚度为1nm的氟化锂(lif),由此形成电子注入层2115。
[0746]
最后,作为用作阴极的第二电极2103,通过蒸镀形成厚度为200nm的铝膜,由此制造本实施例的发光元件3。
[0747]
表5示出经上述步骤而得到的发光元件3的元件结构。
[0748][0749]
在氮气氛的手套箱中,以不使通过上述步骤制造的发光元件2及发光元件3暴露于大气的方式对各发光元件进行密封处理(将密封材料涂敷在元件的周围,在密封时以80℃
进行1小时的热处理)。然后,测定该发光元件2及发光元件3的工作特性。另外,在室温下(在保持为25℃的气氛中)进行测定。
[0750]
图51及图58分别示出发光元件2及发光元件3的电流密度-亮度特性。在图51及图58中,横轴表示电流密度(ma/cm2),而纵轴表示亮度(cd/m2)。另外,图52及图59分别示出发光元件2及发光元件3的电压-亮度特性。在图52及图59中,横轴表示电压(v),而纵轴表示亮度(cd/m2)。另外,图53及图60分别示出发光元件2及发光元件3的亮度-电流效率特性。在图53及图60中,横轴表示亮度(cd/m2),而纵轴表示电流效率(cd/a)。另外,图54及图61分别示出发光元件2及发光元件3的电压-电流特性。在图54及图61中,横轴表示电压(v),而纵轴表示电流(ma)。另外,图55及图62分别示出发光元件2及发光元件3的亮度-色度坐标特性。在图55及图62中,横轴表示亮度(cd/m2),而纵轴表示色度(x坐标及y坐标)。另外,图56及图63分别示出发光元件2及发光元件3的亮度-功率效率特性。在图56及图63中,横轴表示亮度(cd/m2),而纵轴表示功率效率(lm/w)。
[0751]
根据图56及图63可知,发光元件2及发光元件3是效率高的元件。另外,根据图55及图62可知,发光元件2及发光元件3是各亮度上的载流子平衡良好的元件。另外,根据图54及图61可知,发光元件2及发光元件3是驱动电压低且耗电量低的元件。
[0752]
另外,表6示出发光元件2及发光元件3的亮度大约为1000cd/m2时的电压(v)、电流密度(ma/cm2)、cie色度坐标(x,y)、亮度(cd/m2)、电流效率(cd/a)和外部量子效率(%)。
[0753]
[表6]
[0754][0755]
另外,图57及图64分别示出发光元件2及发光元件3的电流密度为2.5ma/cm2时的发射光谱。如图57及图64所示,发光元件2及发光元件3的发射光谱分别在548nm、472nm处具有峰值。
[0756]
另外,如表6所示,发光元件2的亮度为723cd/m2时的cie色度坐标为(x,y)=(0.43,0.56)。另外,发光元件3的亮度为735cd/m2时的cie色度坐标为(x,y)=(0.20,0.35)。由此可知,发光元件2及发光元件3都得到来源于掺杂剂的发光。
[0757]
根据上述可知,本发明的一个方式的发光元件2能够高效地发射黄色波长区的光。另外,本发明的一个方式的发光元件3能够高效地发射蓝色波长区的光。换言之,4,6mdbtp2pm-ii具有高于蓝色光的t1能级,因此,4,6mdbtp2pm-ii优选用作发射可见区域(蓝至红)的光的发光材料的主体材料。
[0758]
实施例14
[0759]
在本实施例中,评价使用本发明的一个方式的在实施例4中合成的由结构式(400)表示的有机化合物的发光元件4、发光元件5、发光元件6及使用4,6-双[3-(9h-咔唑-9-基)苯基]嘧啶(简称:4,6czp2pm)的发光元件7。
[0760]
首先,示出用于本实施例的发光元件4的材料的化学式。
[0761][0762]
参照图50a说明发光元件4。以下,示出本实施例的发光元件4的制造方法。
[0763]
(发光元件4)
[0764]
首先,在衬底2100上通过溅射法形成包含硅或氧化硅的氧化铟-氧化锡化合物(itso),由此形成第一电极2101。另外,所使用的靶材的组成比为in2o3:sno2:sio2=85:10:5[wt.%]。另外,将第一电极2101的厚度设定为110nm,且将其电极面积设定为2mm
×
2mm。这里,第一电极2101是用作发光元件的阳极的电极。
[0765]
接着,作为用来在衬底2100上形成发光元件的预处理,在用水洗涤衬底表面并在200℃下进行焙烧1小时之后,进行uv臭氧处理370秒。
[0766]
然后,将衬底放进到真空蒸镀装置中,其内部被减压到10-4
pa左右。在真空蒸镀装置内的加热室中,在170℃下进行30分钟的真空焙烧,然后使衬底2100冷却30分钟左右。
[0767]
接着,以使形成有第一电极2101的面朝下的方式将形成有第一电极2101的衬底2100固定在设置在真空蒸镀装置内的衬底支架上,并将压力降低到10-4
pa左右,然后在第一
电极2101上共蒸镀dbt3p-ii和氧化钼,从而形成空穴注入层2111。将空穴注入层2111的厚度设定为40nm,将dbt3p-ii与氧化钼的重量比调节为4:2(=dbt3p-ii:氧化钼)。
[0768]
接着,在空穴注入层2111上形成厚度为20nm的bpaflp,由此形成空穴传输层2112。
[0769]
再者,通过共蒸镀在实施例4中合成的4,6-双[3-(二苯并噻吩-4-基)苯基]嘧啶(简称:4,6mdbtp2pm-ii)、4-4
′‑
二(1-萘基)-4
″‑
(9-苯基-9h-咔唑-3-基)三苯胺(简称:pcbnbb)以及(乙酰丙酮合)双(6-叔丁基-4-苯基嘧啶合)铱(iii)(简称:[ir(tbuppm)2(acac)]),在空穴传输层2112上形成第一发光层2113a。这里,将4,6mdbtp2pm-ii、pcbnbb以及[ir(tbuppm)2(acac)]的重量比调节为0.5:0.5:0.05(=4,6mdbtp2pm-ii:pcbnbb:[ir(tbuppm)2(acac)])。另外,将第一发光层2113a的厚度设定为20nm。
[0770]
接着,通过在第一发光层2113a上共蒸镀4,6mdbtp2pm-ii、pcbnbb以及[ir(tbuppm)2(acac)],在第一发光层2113a上形成第二发光层2113b。这里,将4,6mdbtp2pm-ii、pcbnbb以及[ir(tbuppm)2(acac)]的重量比调节为0.8:0.2:0.05(=4,6mdbtp2pm-ii:pcbnbb:[ir(tbuppm)2(acac)])。另外,将第二发光层2113b的厚度设定为20nm。
[0771]
接着,在第二发光层2113b上形成厚度为10nm的4,6mdbtp2pm-ii,由此形成第一电子传输层2114a。
[0772]
接着,在第一电子传输层2114a上形成厚度为20nm的bphen,由此形成第二电子传输层2114b。
[0773]
再者,在第二电子传输层2114b上蒸镀形成厚度为1nm的氟化锂(lif),由此形成电子注入层2115。
[0774]
最后,作为用作阴极的第二电极2103,通过蒸镀形成厚度为200nm的铝膜,由此制造本实施例的发光元件4。
[0775]
表7示出经上述步骤而得到的发光元件4的元件结构。
[0776][0777]
接着,以下示出用于本实施例的发光元件5的材料的化学式。
[0778][0779]
参照图50c说明发光元件5。以下,示出本实施例的发光元件5的制造方法。
[0780]
(发光元件5)
[0781]
首先,在衬底2100上通过溅射法形成包含硅或氧化硅的氧化铟-氧化锡化合物(itso),由此形成第一电极2101。另外,所使用的靶材的组成比为in2o3:sno2:sio2=85:10:5[wt.%]。另外,将第一电极2101的厚度设定为110nm,且将其电极面积设定为2mm
×
2mm。这里,第一电极2101是用作发光元件的阳极的电极。
[0782]
接着,作为用来在衬底2100上形成发光元件的预处理,在用水洗涤衬底表面并在200℃下进行焙烧1小时之后,进行uv臭氧处理370秒。
[0783]
然后,将衬底放进到真空蒸镀装置中,其内部被减压到10-4
pa左右。在真空蒸镀装置内的加热室中,在170℃下进行30分钟的真空焙烧,然后使衬底2100冷却30分钟左右。
[0784]
接着,以使形成有第一电极2101的面朝下的方式将形成有第一电极2101的衬底2100固定在设置在真空蒸镀装置内的衬底支架上,并将压力降低到10-4
pa左右,然后在第一
电极2101上共蒸镀dbt3p-ii和氧化钼,从而形成空穴注入层2111。将空穴注入层2111的厚度设定为40nm,将dbt3p-ii与氧化钼的重量比调节为4:2(=dbt3p-ii:氧化钼)。
[0785]
接着,在空穴注入层2111上共蒸镀9-[3-(9-苯基-9h-芴-9-基)苯基]-9h-咔唑(简称:mczflp)及pccp,由此形成空穴传输层2112。将空穴传输层2112的厚度设定为20nm,将mczflp与pccp的重量比调节为1:1(=mczflp:pccp)。
[0786]
再者,通过共蒸镀在实施例4中合成的4,6mdbtp2pm-ii、pccp及三(2-苯基吡啶-n,c2′
)铱(iii)(简称:[ir(ppy)3]),在空穴传输层2112上形成发光层2113。这里,将4,6mdbtp2pm-ii、pccp以及[ir(ppy)3]的重量比调节为0.8:0.2:0.05(=4,6mdbtp2pm-ii:pccp:[ir(ppy)3])。另外,将发光层2113的厚度设定为40nm。
[0787]
接着,在发光层2113上形成厚度为10nm的4,6mdbtp2pm-ii,由此形成第一电子传输层2114a。
[0788]
接着,在第一电子传输层2114a上形成厚度为20nm的bphen,由此形成第二电子传输层2114b。
[0789]
再者,在第二电子传输层2114b上蒸镀形成厚度为1nm的氟化锂(lif),由此形成电子注入层2115。
[0790]
最后,作为用作阴极的第二电极2103,通过蒸镀形成厚度为200nm的铝膜,由此制造本实施例的发光元件5。
[0791]
表8示出经上述步骤而得到的发光元件5的元件结构。
[0792][0793]
接着,以下示出用于本实施例的发光元件6的材料的化学式。
[0794][0795]
参照图50a说明发光元件6。以下,示出本实施例的发光元件6的制造方法。
[0796]
(发光元件6)
[0797]
首先,在衬底2100上通过溅射法形成包含硅或氧化硅的氧化铟-氧化锡化合物(itso),由此形成第一电极2101。另外,所使用的靶材的组成比为in2o3:sno2:sio2=85:10:5[wt.%]。另外,将第一电极2101的厚度设定为110nm,且将其电极面积设定为2mm
×
2mm。这里,第一电极2101是用作发光元件的阳极的电极。
[0798]
接着,作为用来在衬底2100上形成发光元件的预处理,在用水洗涤衬底表面并在200℃下进行焙烧1小时之后,进行uv臭氧处理370秒。
[0799]
然后,将衬底放进到真空蒸镀装置中,其内部被减压到10-4
pa左右。在真空蒸镀装置内的加热室中,在170℃下进行30分钟的真空焙烧,然后使衬底2100冷却30分钟左右。
[0800]
接着,以使形成有第一电极2101的面朝下的方式将形成有第一电极2101的衬底2100固定在设置在真空蒸镀装置内的衬底支架上,并将压力降低到10-4
pa左右,然后在第一
电极2101上共蒸镀dbt3p-ii和氧化钼,从而形成空穴注入层2111。将空穴注入层2111的厚度设定为30nm,将dbt3p-ii与氧化钼的重量比调节为4:2(=dbt3p-ii:氧化钼)。
[0801]
接着,在空穴注入层2111上形成厚度为20nm的bpaflp,由此形成空穴传输层2112。
[0802]
再者,通过共蒸镀在实施例4中合成的4,6-双[3-(二苯并噻吩-4-基)苯基]嘧啶(简称:4,6mdbtp2pm-ii)、pccp及三[2-甲基-3-(6-叔丁基-4-嘧啶基-κn3)吡啶基-κc](简称:[ir(tbumpypm)3]),在空穴传输层2112上形成第一发光层2113a。这里,将4,6mdbtp2pm-ii、pccp以及[ir(tbumpypm)3]的重量比调节为0.5:0.5:0.05(=4,6mdbtp2pm-ii:pccp:[ir(tbumpypm)3])。另外,将第一发光层2113a的厚度设定为20nm。
[0803]
接着,通过在第一发光层2113a上共蒸镀4,6mdbtp2pm-ii、pccp以及[ir(tbumpypm)3],在第一发光层2113a上形成第二发光层2113b。这里,将4,6mdbtp2pm-ii、pccp以及[ir(tbumpypm)3]的重量比调节为0.8:0.2:0.05(=4,6mdbtp2pm-ii:pccp:[ir(tbumpypm)3])。另外,将第二发光层2113b的厚度设定为20nm。
[0804]
接着,在第二发光层2113b上形成厚度为10nm的4,6mdbtp2pm-ii,由此形成第一电子传输层2114a。
[0805]
接着,在第一电子传输层2114a上形成厚度为15nm的bphen,由此形成第二电子传输层2114b。
[0806]
再者,在第二电子传输层2114b上蒸镀形成厚度为1nm的氟化锂(lif)膜,由此形成电子注入层2115。
[0807]
最后,作为用作阴极的第二电极2103,通过蒸镀形成厚度为200nm的铝膜,由此制造本实施例的发光元件6。
[0808]
表9示出经上述步骤而得到的发光元件6的元件结构。
[0809][0810]
接着,以下示出用于本实施例的发光元件7的材料的化学式。
[0811][0812]
参照图50a说明发光元件7。以下,示出本实施例的发光元件7的制造方法。
[0813]
(发光元件7)
[0814]
首先,在衬底2100上通过溅射法形成包含硅或氧化硅的氧化铟-氧化锡化合物(itso),由此形成第一电极2101。另外,所使用的靶材的组成比为in2o3:sno2:sio2=85:10:5[wt.%]。另外,将第一电极2101的厚度设定为110nm,且将其电极面积设定为2mm
×
2mm。这里,第一电极2101是用作发光元件的阳极的电极。
[0815]
接着,作为用来在衬底2100上形成发光元件的预处理,在用水洗涤衬底表面并在200℃下进行焙烧1小时之后,进行uv臭氧处理370秒。
[0816]
然后,将衬底放进到真空蒸镀装置中,其内部被减压到10-4
pa左右。在真空蒸镀装置内的加热室中,在170℃下进行30分钟的真空焙烧,然后使衬底2100冷却30分钟左右。
[0817]
接着,以使形成有第一电极2101的面朝下的方式将形成有第一电极2101的衬底2100固定在设置在真空蒸镀装置内的衬底支架上,并将压力降低到10-4
pa左右,然后在第一
电极2101上共蒸镀dbt3p-ii和氧化钼,从而形成空穴注入层2111。将空穴注入层2111的厚度设定为40nm,将dbt3p-ii与氧化钼的重量比调节为4:2(=dbt3p-ii:氧化钼)。
[0818]
接着,在空穴注入层2111上形成厚度为20nm的mczflp,由此形成空穴传输层2112。
[0819]
再者,通过共蒸镀4,6-双[3-(9h-咔唑-9-基)苯基]嘧啶(简称:4,6mczp2pm)、pccp以及双[2-甲基-3-(6-叔丁基-4-嘧啶基-κn3)吡啶基-κc](2,4-戊二酮合-κ2o,o
′
)铱(iii))(简称:[ir(tbumpypm)2(acac)]),在空穴传输层2112上形成第一发光层2113a。这里,将4,6mczp2pm、pccp以及[ir(tbumpypm)2(acac)]的重量比调节为0.8:0.2:0.05(=4,6mczp2pm:pccp:[ir(tbumpypm)2(acac)])。另外,将第一发光层2113a的厚度设定为20nm。
[0820]
接着,通过在第一发光层2113a上共蒸镀4,6mczp2pm、以及[ir(tbumpypm)2(acac)],在第一发光层2113a上形成第二发光层2113b。这里,将4,6mczp2pm、与[ir(tbumpypm)2(acac)]的重量比调节为1:0.05(=4,6mczp2pm:[ir(tbumpypm)2(acac)])。另外,将第二发光层2113b的厚度设定为20nm。
[0821]
接着,在第二发光层2113b上形成厚度为10nm的4,6mczp2pm,由此形成第一电子传输层2114a。
[0822]
接着,在第一电子传输层2114a上形成厚度为20nm的bphen,由此形成第二电子传输层2114b。
[0823]
再者,在第二电子传输层2114b上蒸镀形成厚度为1nm的氟化锂(lif)膜,由此形成电子注入层2115。
[0824]
最后,作为用作阴极的第二电极2103,通过蒸镀形成厚度为200nm的铝膜,由此制造本实施例的发光元件7。
[0825]
表10示出经上述步骤而得到的发光元件7的元件结构。
[0826][0827]
在氮气氛的手套箱中,以不使通过上述步骤制造的发光元件4至发光元件7暴露于大气的方式对各发光元件进行密封处理(将密封材料涂敷在元件的周围,在密封时以80℃
进行1小时的热处理)。然后,测定该发光元件4至发光元件7的工作特性。另外,在室温下(在保持为25℃的气氛中)进行测定。
[0828]
图65、图72、图79及图86分别示出发光元件4、发光元件5、发光元件6及发光元件7的电流密度-亮度特性。在图65、图72、图79及图86中,横轴表示电流密度(ma/cm2),而纵轴表示亮度(cd/m2)。另外,图66、图73、图80及图87分别示出发光元件4、发光元件5、发光元件6及发光元件7的电压-亮度特性。在图66、图73、图80及图87中,横轴表示电压(v),而纵轴表示亮度(cd/m2)。另外,图67、图74、图81及图88分别示出发光元件4、发光元件5、发光元件6及发光元件7的亮度-电流效率特性。在图67、图74、图81及图88中,横轴表示亮度(cd/m2),而纵轴表示电流效率(cd/a)。另外,图68、图75、图82及图89分别示出发光元件4、发光元件5、发光元件6及发光元件7的电压-电流特性。在图68、图75、图82及图89中,横轴表示电压(v),而纵轴表示电流(ma)。另外,图69、图76、图83及图90分别示出发光元件4、发光元件5、发光元件6及发光元件7的亮度-色度坐标特性。在图69、图76、图83及图90中,横轴表示亮度(cd/m2),而纵轴表示色度(x坐标及y坐标)。另外,图70、图77、图84及图91分别示出发光元件4、发光元件5、发光元件6及发光元件7的亮度-功率效率特性。在图70、图77、图84及图91中,横轴表示亮度(cd/m2),而纵轴表示功率效率(lm/w)。
[0829]
根据图70、图77、图84及图91可知,发光元件4至发光元件7是效率高的元件。另外,根据图69、图76、图83及图90可知,发光元件4至发光元件7是各亮度上的载流子平衡良好的元件。另外,根据图68、图75、图82及图89可知,发光元件4至发光元件7是驱动电压低且耗电量低的元件。
[0830]
另外,表11示出发光元件4至发光元件7的亮度大约为1000cd/m2时的电压(v)、电流密度(ma/cm2)、cie色度坐标(x,y)、亮度(cd/m2)、电流效率(cd/a)和外部量子效率(%)。
[0831]
[表11]
[0832][0833]
另外,图71、图78、图85及图92分别示出发光元件4至发光元件7的电流密度为2.5ma/cm2时的发射光谱。如图71、图78、图85及图92所示,发光元件4至发光元件7的发射光谱分别在548nm、513nm、493nm、508nm处具有峰值。
[0834]
如上所述,可知获得了来自各掺杂剂材料(铱配合物)的发光。并且,由于本发明的
一个方式的第二化合物的包含嘧啶骨架的有机化合物具有高t1能级,所以可知能够将其用作发射蓝绿光或比蓝绿色更长的波长的光的磷光材料的主体材料。
[0835]
另外,如表11所示,发光元件4的亮度为694cd/m2时的cie色度坐标为(x,y)=(0.43,0.56)。另外,发光元件4的亮度为694cd/m2时的电压、电流密度、电流效率及外部量子效率分别是2.7v、0.9ma/cm2、79cd/a及22%。另外,发光元件5的亮度为954cd/m2时的cie色度坐标为(x,y)=(0.33,0.62)。另外,发光元件5的亮度为954cd/m2时的电压、电流密度、电流效率及外部量子效率分别是3.1v、1.4ma/cm2、69cd/a及20%。另外,发光元件6的亮度为797cd/m2时的cie色度坐标为(x,y)=(0.25,0.52)。另外,发光元件6的亮度为797cd/m2时的电压、电流密度、电流效率及外部量子效率分别是2.9v、1.3ma/cm2、60cd/a及24%。另外,发光元件7的亮度为810cd/m2时的cie色度坐标为(x,y)=(0.30,0.61)。另外,发光元件7的亮度为810cd/m2时的电压、电流密度、电流效率及外部量子效率分别是3.3v、1.1ma/cm2、77cd/a及24%。
[0836]
如上所述,可知:本发明的一个方式的发光元件4至发光元件7具有驱动电压低且载流子平衡良好的特性。
[0837]
由于本发明的一个方式的第二化合物的包含嘧啶骨架的有机化合物具有高电子传输性,所以可知当将其用于电子传输层时可以获得驱动电压低的元件。
[0838]
另外,如发光元件4至发光元件7的说明中所示,当将本发明的一个方式的第二化合物的包含嘧啶骨架的有机化合物及空穴传输性高的化合物(pcbnbb或pccp)用于发光层时,能够将电子及空穴高效地注入到掺杂剂(铱配合物),从而可以获得效率高且驱动电压低的元件。
[0839]
另外,如发光元件5的说明中所示,当将空穴传输性高的材料(pccp)及t1能级高的材料(mczflp)用于空穴传输层时,空穴高效地被注入到发光层且能够抑制产生在发光层中的激发能量传到空穴传输层,从而可以获得驱动电压更低且效率更高的元件。
[0840]
接着,对上述发光元件4及发光元件5进行可靠性试验的评价。图93至图96示出可靠度试验的结果。
[0841]
在可靠性试验中,在将初始亮度设定为5000cd/m2并电流密度恒定的条件下驱动发光元件4及发光元件5。图93及图95示出其结果。横轴表示元件的驱动时间(h),而纵轴表示起始亮度为100%时的归一化亮度(%)。根据图93可知,大约300小时后,发光元件4的归一化亮度降低到87%。另外,根据图95可知,大约420小时后,发光元件5的归一化亮度降低到83%。
[0842]
接着,在可靠性试验中,在将初始亮度设定为5000cd/m2并电流密度恒定的条件下测定发光元件4及发光元件5的电压的随时间变化。图94及图96示出其结果。横轴表示元件的驱动时间(h),而纵轴表示电压(v)。根据图94及图96可知,发光元件4及发光元件5的电压的随时间上升的程度小。
[0843]
如上所述可知,发光元件4及发光元件5是长使用寿命的元件。
[0844]
(参考例1)
[0845]
对上述实施例12及实施例13中使用的(乙酰丙酮根)双(6-叔丁基-4-苯基嘧啶基)铱(iii)(简称:[ir(tbuppm)2(acac)])的合成方法进行具体的说明。另外,以下示出[ir(tbuppm)2(acac)]的结构。
[0846][0847]
《步骤1:4-叔丁基-6-苯基嘧啶(简称:htbuppm)的合成》
[0848]
首先,在装有回流管的茄形烧瓶中,放入22.5g 4,4-二甲基-1-戊基苯-1,3-二酮和50g甲酰胺,用氮气置换烧瓶内部的空气。加热该反应容器来将反应溶液回流5小时。然后,对氢氧化钠水溶液注入该溶液,使用二氯甲烷萃取有机层。用水、饱和盐水洗涤得到的有机层,并用硫酸镁进行干燥。过滤干燥后的溶液。将该溶液的溶剂蒸馏而去除,然后将所得到的残渣通过硅胶柱色谱法使用己烷:乙酸乙酯=10:1(体积比)作为展开剂来纯化。结果,得到嘧啶衍生物htbuppm(无色油状物,收率14%)。下记(m-1)示出步骤1的合成方案。
[0849][0850]
《步骤2:二-μ-氯-双[双(6-叔丁基-4-苯基嘧啶根)铱(iii)](简称:[ir(tbuppm)2cl]2)的合成》
[0851]
接着,在装有回流管的茄形烧瓶中,放入15ml 2-乙氧基乙醇、5ml水、1.49g在上述步骤1得到的htbuppm和1.04g氯化铱水合物(ircl3·
h2o),用氩气置换空气。然后,照射1小时的微波(2.45ghz,100w)来进行反应。在蒸馏而去除该溶液的溶剂之后,将所得到的残渣使用乙醇抽滤,并洗涤得到双核配合物[ir(tbuppm)2cl]2(黄绿色粉末,收率73%)。下记(m-2)示出步骤2的合成方案。
[0852][0853]
《步骤3:(乙酰丙酮根)双(6-叔丁基-4-苯基嘧啶根)铱(iii)(简称:[ir(tbuppm)2(acac)])的合成》
[0854]
再者,在装有回流管的茄形烧瓶中,放入40ml 2-乙氧基乙醇、1.61g在上述步骤2得到的双核配合物[ir(tbuppm)2cl]2、0.36g乙酰丙酮和1.27g碳酸钠,用氩气置换烧瓶内部的空气。然后,照射1小时的微波(2.45ghz,120w)来进行反应。蒸馏而去除溶剂,将得到的残渣使用乙醇抽滤,并使用水、乙醇洗涤该残渣。将该固体溶解于二氯甲烷中,通过依次层叠硅藻土(日本和光纯药工业株式会社、目录号码:531-16855)、矾土、硅藻土的过滤助剂进行过滤。通过利用二氯甲烷和己烷的混合溶剂使通过蒸馏去除溶剂来得到的固体再结晶,得到作为目的物的黄色粉末(收率68%)。下记(m-3)示出步骤3的合成方案。
[0855][0856]
利用核磁共振分光法(1h nmr)对通过上述步骤3得到的黄色粉末进行测量。以下示出测定数据。由测量结果可知得到了[ir(tbuppm)2(acac)]。
[0857]
以下示出所得到的物质的1h nmr数据。
[0858]1h nmr.δ(cdcl3):1.50(s,18h),1.79(s,6h),5.26(s,1h),6.33(d,2h),6.77(t,2h),6.85(t,2h),7.70(d,2h),7.76(s,2h),9.02(s,2h)。
[0859]
(参考例2)
[0860]
对上述实施例13使用的三[3-甲基-1-(2-甲基苯基)-5-苯基-1h-1,2,4-三唑合(triazolato)]铱(iii)(简称:[ir(mptz1-mp)3])的合成方法进行具体的说明。以下示出[ir(mptz1-mp)3]的结构。
[0861][0862]
《步骤1:n-(1-乙氧基乙烯基)苯甲酰胺的合成》
[0863]
首先,在500ml的三口烧瓶中,放入15.5g乙基乙酰亚胺盐酸盐、150ml甲苯、31.9g三乙胺(et3n),并在室温下搅拌10分钟。使用滴液漏斗将17.7g苯甲酰氯和30ml甲苯的混合溶剂50ml滴加到该混合物中,并在室温下搅拌24小时。在经过预定时间之后,对反应混合物进行抽滤,并使用甲苯洗涤固体。浓缩所得到的滤液来得到n-(1-乙氧基乙烯基)苯甲酰胺(红色油状物,收率82%)。下记(n-1)示出步骤1的合成方案。
[0864][0865]
《步骤2:3-甲基-1(2-甲基苯基)-5-苯基-1h-1,2,4-三唑(简称:hmptz1-mp)的合成》
[0866]
接着,在300ml的茄形烧瓶中,8.68g邻-甲苯基肼盐酸盐、100ml四氯化碳、35ml三乙胺(et3n)放入,并在室温下搅拌1小时。在预定时间之后,对该混合物添加通过上述步骤1得到的8.72g n-(1-乙氧基乙烯基)苯甲酰胺,且在室温下搅拌24小时。在经过预定的时间之后,对该混合物添加水,使用氯仿萃取水层。使用饱和食盐水洗涤该有机层,并添加无水硫酸镁进行干燥。对所得到的混合物进行自然过滤,并浓缩滤液来得到油状物。作为展开剂,使用二氯甲烷。浓缩所得到的馏分来得到3-甲基-1(2-甲基苯基)-5-苯基-1h-1,2,4-三唑(简称:hmptz1-mp)(橙色油状物,收率84%)。下记(n-2)示出步骤2的合成方案。
[0867][0868]
《步骤3:三[3-甲基-1-(2-甲基苯基)-5-苯基-1h-1,2,4-三唑(triazolato)]铱(iii)(简称:[ir(mptz1-mp)3])的合成》
[0869]
接着,在安装有三通旋塞的反应容器中,放入通过上述步骤2得到的2.71g配体hmptzl-mp及1.06g三(乙酰丙酮)铱(iii)。用氩气置换反应容器的空气,并以250℃进行48小时的加热进行反应。将该反应混合物溶解于二氯甲烷中,通过硅胶柱色谱法进行纯化。作为展开剂,首先使用二氯甲烷,然后二氯甲烷:乙酸乙酯=10:1(v/v)的混合溶剂。浓缩所获得的馏分来得到固体。使用乙酸乙酯洗涤该固体,然后使用二氯甲烷和乙酸乙酯的混合溶剂使其再结晶,从而得到本发明的一个方式的有机金属配合物[ir(mptz1-mp)3](黄色粉末,收率35%)。下记(n-3)示出步骤3的合成方案。
[0870][0871]
利用核磁共振分光法(1h-nmr)对通过上述步骤3得到的黄色粉末进行测量。由测量结果可知得到了[ir(mptz1-mp)3]。
[0872]
以下示出所得到的物质的1h nmr数据。
[0873]1h nmr.δ(cdcl3):1.94-2.21(m、18h)、6.47-6.76(m、12h)、7.29-7.52(m、12h)。
[0874]
参考例3
[0875]
对上述实施例14所使用的三[2-甲基-3-(6-叔丁基-4-嘧啶基-κn3)吡啶-κc]铱(iii)(简称:[ir(tbumpypm)3])的合成方法进行说明。以下,示出[ir(tbumpypm)3]的结构。
[0876][0877]
《步骤1:4-羟基-6-叔丁基嘧啶的合成》
[0878]
首先,对100ml的三口烧瓶放入加入7.2g甲脒乙酸盐、7.5g甲醇钠及70ml甲醇,并对该混合溶液加入10g甲基4,4-二甲基氧代戊酸乙酯,在室温下搅拌24小时。在经过预定的时间之后,对该混合溶液加入水17ml与乙酸7.2ml的混合溶液,在室温下进行搅拌。对该混合物进行浓缩,将得到的残渣溶解于水,并使用乙酸乙酯进行萃取。用饱和食盐水洗涤得到的萃取溶液,并对有机层加入无水硫酸镁进行干燥。对所得到的混合物进行自然过滤,并且浓缩滤液,以得到固体。使用乙酸乙酯对该固体进行洗涤得到4-羟基-6-叔丁基嘧啶(白色固体,收率49%)。下记(p-1)示出步骤1的合成方案。
[0879][0880]
《步骤2:4-氯-6-叔丁基嘧啶的合成》
[0881]
接着,将4.7g通过上述步骤1得到的4-羟基-6-叔丁基嘧啶、14ml磷酰氯加入50ml的三口烧瓶中,并进行1.5小时的加热回流。在回流后,在减压下蒸馏去除磷酰氯。将得到的残渣溶解于二氯甲烷中,并利用水、饱和碳酸氢钠水溶液进行洗涤。对得到的有机层加入无水硫酸镁进行干燥。对该混合物进行自然过滤,并浓缩滤液,以得到固体。通过硅胶柱色谱法对该固体进行纯化。作为展开剂,使用己烷:乙酸乙酯=10:1(v/v)的混合溶剂。通过浓缩得到的馏分得到4-氯-6-叔丁基嘧啶(白色固体,收率78%)。下记(p-2)示出步骤2的合成方案。
[0882][0883]
《步骤3:4-(2-甲基吡啶-3-基)-6-叔丁基嘧啶(简称:htbumpypm)的合成》
[0884]
接着,将2.0g通过上述步骤2得到的4-氯-6-叔丁基嘧啶、3.0g2-甲基吡啶-3-硼酸频哪醇酯、1m醋酸钾水溶液17ml、1m碳酸钠水溶液17ml及40ml乙腈放入100ml的圆底烧瓶中,用氩气置换烧瓶内部的空气。对该混合物加入0.78g四(三苯基膦)钯(0),并在100℃、100w的条件下进行1小时的微波照射使其反应。使用乙酸乙酯对该反应混合物进行萃取,并使用饱和食盐水进行洗涤。对得到的萃取溶液加入无水硫酸镁进行干燥,并对混合物进行自然过滤得到滤液。将得到的滤液溶解于乙酸乙酯与己烷的混合溶剂中,通过硅藻土、矾土、硅藻土进行过滤。通过硅胶柱色谱法对得到的滤液进行纯化。作为展开剂使用己烷:乙酸乙酯=3:2(v/v)的混合溶剂。将所得到的馏分浓缩获得油状物。将该油状物溶解于己烷与乙酸乙酯的混合溶剂中,并使其通过以硅藻土、矾土、硅藻土的顺序层叠的助滤剂进行过滤。浓缩得到的滤液得到4-(2-甲基吡啶-3-基)-6-叔丁基嘧啶(简称:htbumpypm)(淡黄色油状物,收率92%)。下记(p-3)示出步骤3的合成方案。
[0885][0886]
《步骤4:三[2-甲基-3-(6-叔丁基-4-嘧啶基-κn3)吡啶-κc]铱(iii)(简称[ir(tbumpypm)3])的合成》
[0887]
接着,将3.31g通过上述步骤3得到的配体htbumpypm、1.42g三(乙酰丙酮合)铱(iii)放入安装有三通旋塞的反应容器中,并用氩气置换反应容器内部的空气。然后,以250℃的温度加热50.5小时,进行反应。利用使用乙酸乙酯:甲醇=4:1的展开剂的快速柱色谱法对得到的残渣进行纯化。通过利用二氯甲烷与己烷的混合溶剂使通过蒸馏去除馏分溶剂得到的固体再结晶两次,得到[ir(tbumpypm)3](黄褐色粉末,收率22%)。下记(p-4)示出步骤4的合成方案。
[0888][0889]
利用核磁共振分光法(1h-nmr)对通过上述步骤4得到的黄褐色粉末进行测量。由测量结果可知得到了[ir(tbumpypm)3]。
[0890]
以下示出得到的物质的1h nmr数据。
[0891]1h-nmr.δ(cdcl3):1.41(s,27h),2.94(s,9h),6.64(d,3h),7.70(d,3h),8.12(s,3h),8.24(s,3h)。
[0892]
参考例4
[0893]
对上述实施例14所使用的三[2-甲基-3-(6-叔丁基-4-嘧啶基-κn3)吡啶基-κc](2,4-戊二酮合-κ20,0
′
)铱(iii)(简称:[ir(tbumpypm)2(acac)])的合成方法进行说明。以下,示出[ir(tbumpypm)2(acac)]的结构。
[0894][0895]
《步骤1:4-羟基-6-叔丁基嘧啶的合成》
[0896]
首先,对100ml的三口烧瓶放入7.2g甲脒乙酸盐、7.5g甲醇钠及70ml甲醇,并对该混合溶液加入10g甲基4,4-二甲基氧代戊酸乙酯,在室温下搅拌24小时。在经过预定的时间之后,对该混合溶液加入水17ml与乙酸7.2ml的混合溶液,在室温下进行搅拌。对该混合物进行浓缩,将得到的残渣溶解于水,并使用乙酸乙酯进行萃取。用饱和食盐水洗涤得到的萃取溶液,并对有机层加入无水硫酸镁进行干燥。对所得到的混合物进行自然过滤,并浓缩滤液,以得到固体。使用乙酸乙酯对该固体进行洗涤得到4-羟基-6-叔丁基嘧啶(白色固体、收率49%)。下记(q-1)示出步骤1的合成方案。
[0897][0898]
《步骤2:4-氯-6-叔丁基嘧啶的合成》
[0899]
接着,将4.7g通过上述步骤1得到的4-羟基-6-叔丁基嘧啶及14ml磷酰氯放入50ml的三口烧瓶中,并进行1.5小时的加热回流。在回流后,在减压下蒸馏去除磷酰氯。将得到的残渣溶解于二氯甲烷,并利用水、饱和碳酸氢钠水溶液进行洗涤。对得到的有机层加入无水硫酸镁进行干燥。对该混合物进行自然过滤,并且浓缩滤液,以得到固体。通过硅胶柱色谱法使该固体纯化。作为展开剂,使用己烷:乙酸乙酯=10:1(v/v)的混合溶剂。浓缩得到的馏分得到4-氯-6-叔丁基嘧啶(白色固体,收率78%)。下记(q-2)示出步骤2的合成方案。
[0900][0901]
《步骤3:4-(2-甲基吡啶-3-基)-6-叔丁基嘧啶(简称:htbumpypm)的合成》
[0902]
接着,将2.0g通过上述步骤2得到的4-氯-6-叔丁基嘧啶、3.0g2-甲基吡啶-3-硼酸频哪醇酯、1m醋酸钾水溶液17ml、1m碳酸钠水溶液17ml及40ml乙腈加入100ml的圆底烧瓶中,用氩气置换烧瓶内部的空气。对该混合物加入0.78g四(三苯基膦)钯(0),并在100℃、100w的条件下进行1小时的微波照射使其反应。使用乙酸乙酯对该反应混合物进行萃取,并使用饱和食盐水进行洗涤。对得到的萃取溶液加入无水硫酸镁进行干燥,并对混合物进行自然过滤得到滤液。将得到的滤液溶解于乙酸乙酯与己烷的混合溶剂中,通过硅藻土、矾土、硅藻土进行过滤。通过硅胶柱色谱法对得到的滤液进行纯化。作为展开剂使用己烷:乙酸乙酯=3:2(v/v)的混合溶剂。将所得到的馏分浓缩获得油状物。将该油状物溶解于己烷与乙酸乙酯的混合溶剂中,并使其通过以硅藻土、矾土、硅藻土的顺序层叠的助滤剂进行过滤。浓缩得到的滤液得到4-(2-甲基吡啶-3-基)-6-叔丁基嘧啶(简称:htbumpypm)(淡黄色油状物、收率92%)。下记(q-3)示出步骤3的合成方案。
[0903][0904]
《步骤4:二-μ-氯-四[2-甲基-3-(6-叔丁基-4-嘧啶基-κn3)吡啶-κc]二铱(iii)(简称:[ir(tbumpypm)2cl]2)的合成》
[0905]
接着,将2.0g通过上述步骤3得到的配体htbumpypm、1.1g氯化铱、21ml 2-乙氧基
乙醇、7ml水放入50ml的茄形烧瓶中,用氩气置换烧瓶内部的空气。通过在100℃、100w的条件下对该反应容器进行1小时的微波照射来进行加热并进行反应。在经过预定的时间之后,浓缩得到的反应溶液得到双核配合物[ir(tbumpypm)2cl]2(橙色油状物,收率100%)。下记(q-4)示出步骤4的合成方案。
[0906][0907]
《步骤5:双[2-甲基-3-(6-叔丁基-4-嘧啶基-κn3)吡啶-κc](2,4-戊二酮根-κ20,0
′
)铱(iii)(简称[ir(tbumpypm)2(acac)])的合成》
[0908]
接着,将2.5g通过上述步骤4得到的双核配合物[ir(tbumpypm)2cl]2、1.9g碳酸钠、0.55g乙酰丙酮、20ml 2-乙氧基乙醇放入100ml的圆底烧瓶中,用氩气置换烧瓶内部的空气。通过在100℃、120w的条件下对该反应容器进行1小时的微波照射进行反应。在经过预定的时间之后,对得到的反应混合物进行浓缩并加入乙醇,有沉淀析出。对该混合物进行抽滤,并使用乙醇洗涤固体得到[ir(tbumpypm)2(acac)](黄色粉末,收率24%)。下记(q-5)示出步骤5的合成方案。
[0909][0910]
利用核磁共振分光法(1h-nmr)对通过上述步骤5得到的黄色粉末进行测量。由测量结果可知得到了[ir(tbumpypm)2(acac)]。
[0911]
以下示出得到的物质的1h nmr数据。
[0912]1h-nmr.δ(cdcl3):1.52(s,18h),1.81(s,6h),2.89(s,6h),5.3(s,1h),6.09(d,2h),6.54(d,2h),8.08(s,2h),9.06(d,2h)。
[0913]
参考例5
[0914]
对实施例14所使用的9-[3-(9-苯基-9h-芴-9-基)苯基]-9h-咔唑(简称:mczflp)的合成方法进行说明。
[0915]
《9-[3-(9-苯基-9h-芴-9-基)苯基]-9h-咔唑(简称:mczflp)的合成法》
[0916]
以下示出mczflp的结构。
[0917][0918]
将4.9g(12.4mmol)9-(3-溴苯基)-9-苯基芴、2.1g(12.4mmol)咔唑及3.6g(37.2mmol)叔丁醇钠放入100ml的三口烧瓶中,用氩气置换烧瓶内部的空气。对该混合物加入31.0ml二甲苯、0.2ml三(叔丁基)膦的10%己烷溶液及48.1mg(0.1mmol)双(二亚苄基丙酮)钯(0),并以140℃搅拌3.5小时。搅拌后,加入47.7mg(0.1mmol)双(二亚苄基丙酮)钯(0)
与0.6ml三(叔丁基)膦的10%己烷溶液,搅拌1.5小时。
[0919]
搅拌后,加入70ml乙酸乙酯、150ml甲苯进行加热,并通过硅酸镁、硅藻土、矾土进行抽滤得到滤液。对得到的滤液进行浓缩,以得到固体。利用硅胶柱色谱法(展开溶剂为己烷:甲苯=7:3)对该固体进行纯化,得到目的物的白色固体。利用甲苯与己烷的混合剂使得到的白色固体再结晶,以46%的收率得到2.7g的目的物白色固体。
[0920]
利用梯度升华法纯化得到的1.5g白色固体。在压力为2.7pa且氩流量为5.0ml/min的条件下,以203℃对固体进行加热。纯化之后,以93%的收率得到1.4g的目的物的白色固体。下记(r-1)示出上述合成法的合成方案。
[0921][0922]
利用核磁共振法(1h nmr)对通过上述合成方案(r-1)得到的化合物进行测量。由测量结果可知得到mczflp。
[0923]
以下示出得到的物质的1h nmr数据。
[0924]1h nmr(cdcl3,500mhz):δ=7.19-7.49(m,21h),7.77(d,j=7.5hz,2h),8.10(d,j=7.0hz,2h)。
[0925]
符号说明
[0926]
100 衬底
[0927]
101 第一电极
[0928]
102 el层
[0929]
103 第二电极
[0930]
111 空穴注入层
[0931]
112 空穴传输层
[0932]
113 发光层
[0933]
114 电子传输层
[0934]
115 电子注入层
[0935]
116 电荷产生层
[0936]
201 第一电极
[0937]
202 第二电极
[0938]
203 el层
[0939]
204 发光层
[0940]
205 磷光化合物
[0941]
206 第一有机化合物
[0942]
207 第二有机化合物
[0943]
301 第一电极
[0944]
302(1) 第一el层
[0945]
302(2) 第二el层
[0946]
304 第二电极
[0947]
305 电荷产生层
[0948]
401 源极侧驱动电路
[0949]
402 像素部
[0950]
403 栅极侧驱动电路
[0951]
404 密封基板
[0952]
405 密封材料
[0953]
407 空间
[0954]
408 引线
[0955]
410 元件衬底
[0956]
411 开关tft
[0957]
412 电流控制tft
[0958]
413 第一电极
[0959]
414 绝缘物
[0960]
416 发光层
[0961]
417 第二电极
[0962]
418 发光元件
[0963]
423 n沟道型tft
[0964]
424 p沟道型tft
[0965]
450r 第一发光元件
[0966]
450g 第二发光元件
[0967]
450b 第三发光元件
[0968]
451 反射电极
[0969]
452 半透过
·
半反射电极
[0970]
453a 第一透明导电层
[0971]
453b 第二透明导电层
[0972]
454b 第一发光层
[0973]
454g 第二发光层
[0974]
454r 第三发光层
[0975]
455 el层
[0976]
501 衬底
[0977]
502 第一电极
[0978]
503 第二电极
[0979]
504 el层
[0980]
505 绝缘层
[0981]
506 分隔层
[0982]
611 框体
[0983]
612 支撑台
[0984]
613 显示部
[0985]
614 扬声器部
[0986]
615 视频输入端子
[0987]
621 主体
[0988]
622 框体
[0989]
623 显示部
[0990]
624 键盘
[0991]
625 外部连接端口
[0992]
626 定位装置
[0993]
631 主体
[0994]
632 框体
[0995]
633 显示部
[0996]
634 声音输入部
[0997]
635 声音输出部
[0998]
636 操作键
[0999]
637 外部连接端口
[1000]
638 天线
[1001]
641 主体
[1002]
642 显示部
[1003]
643 框体
[1004]
644 外部连接端口
[1005]
645 遥控接收部
[1006]
646 图像接收部
[1007]
647 电池
[1008]
648 声音输入部
[1009]
649 操作键
[1010]
650 取景器
[1011]
701 框体
[1012]
702 液晶层
[1013]
703 背光灯
[1014]
704 框体
[1015]
705 驱动器ic
[1016]
706 端子
[1017]
801 框体
[1018]
802 光源
[1019]
901 照明装置
[1020]
902 电视装置
[1021]
1000 衬底
[1022]
1010 第一电极
[1023]
1020 el层
[1024]
1030 第二电极
[1025]
1100 衬底
[1026]
1101 第一电极
[1027]
1103 第二电极
[1028]
1110 空穴注入层
[1029]
1111 空穴注入层
[1030]
1112 空穴传输层
[1031]
1113a 第一发光层
[1032]
1113b 第二发光层
[1033]
1114a 第一电子传输层
[1034]
1114b 第二电子传输层
[1035]
1115 电子注入层
[1036]
1120 空穴传输层
[1037]
1130 发光层
[1038]
1140 电子传输层
[1039]
1150 电子注入层
[1040]
2100 衬底
[1041]
2101 第一电极
[1042]
2103 第二电极
[1043]
2111 空穴注入层
[1044]
2112 空穴传输层
[1045]
2113a 第一发光层
[1046]
2113b 第二发光层
[1047]
2114 电子传输层
[1048]
2114a 第一电子传输层
[1049]
2114b 第二电子传输层
[1050]
2115 电子注入层
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。