1.本发明属于高分子技术领域,具体涉及一种聚酯酰亚胺共聚物及其制备方法和应用。
背景技术:
2.聚酰亚胺由于其优异的热稳定性、化学稳定性和介电性能,在航空航天、电子电器、汽车和化工机械等领域都得到了广泛的应用。传统的聚酰亚胺薄膜由于二酐单体和二胺单体在聚合过程中,会产生电子转移络合物,影响产品薄膜的光学性能,从而进一步限制了聚酰亚胺在光学领域的应用。
3.聚酯酰亚胺是指在聚酰亚胺主链中引入酯键,重复单元以酯基和酰亚胺基为结构特征基团的一类聚合物,独特的分子结构使其有利于协调统筹传统聚酯和聚酰亚胺两者的优势,具有优异的热稳定性、绝缘性、电磁屏蔽性、耐溶剂性以及出色的机械性能。现有技术中报道的以叔丁基对苯二酚双(偏苯三酸酐)为原料制备的聚酯酰亚胺薄膜在400nm处的透过率仅为58.7%,玻璃化转变温度为234℃,热性能和光学性能有待进一步改善(hasegawa m,ishigami t,ishii j.optically transparent aromatic poly(ester imide)s with low coefficients of thermal expansion(1).self-orientation behavior during solution casting process and substituent effect[j].polymer,2015,74:1-15.)。
[0004]
另外,公开号为cn114656636a的中国专利文献公开了一种聚酯酰亚胺,该发明在保护气氛下,将酸类单体、醇类单体、酸酐类单体和胺类单体混合,并调整醇类单体和酸类单体的摩尔比为2.5-4.0:1,在催化剂作用下进行反应,不添加溶剂,制备得到聚酯酰亚胺,但该聚酯酰亚胺的玻璃化转变温度在117-178℃之间,热学性能有待提高;公开号为cn106810695a的中国专利文献公开了一种芳香族聚酯酰亚胺,该方法在熔融加工过程中,预先制备的非对称的含酰亚胺键的芳香族二酚单体与间位全芳香族二酸单体缩聚形成不易结晶的聚酯酰亚胺分子链,而ab型全芳香族单体形成的聚酯分子链易结晶,两者通过间位全芳香族二羰基连接,分子链间存在动态的酯交换反应,有利于建立较强的分子链间化学键作用力;在后续冷却过程中,聚酯分子链迅速结晶,并且均匀分散在不易结晶的含聚酯酰亚胺分子链中,从而得到一种拉伸强度和tg自增强的芳香族聚酯酰亚胺。但该方法较为复杂,对参数的要求高。
技术实现要素:
[0005]
为了提高现有技术中聚酯酰亚胺薄膜的热性能和光学性能,本发明提供了一种聚酯酰亚胺共聚物,利用该聚酯酰亚胺共聚物制得的薄膜玻璃化转变温度高,热膨胀系数低,光学性能好。
[0006]
具体采用的技术方案如下:
[0007]
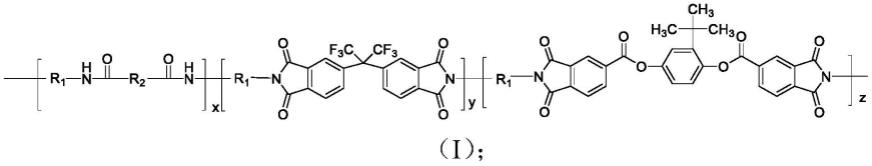
一种聚酯酰亚胺共聚物,结构式如式(ⅰ)所示:
[0008][0009]
其中,x,y,z均为1-100的整数;
[0010]
r1选自式(ⅱ)所示基团中的任一种:
[0011][0012]
r2选自式(ⅲ)所示基团中的任一种:
[0013][0014]
优选的,r1为r2为
[0015]
本发明将酰胺结构引入到聚酯酰亚胺主链中,在分子链间形成氢键,增强链的刚性和线性度,并增强分子链间的作用力,从而提高了聚酯酰亚胺的玻璃化转变温度,降低了其热膨胀系数;另外引入强吸电子基团—三氟甲基和大体积侧基—叔丁基,可以提升其光学性能。
[0016]
优选的,x和z的比例为0.1-7:1;当酰胺链段和聚酯链段的比例在上述范围内时,产物聚酯酰亚胺表现出更加优异的热性能和光学性能。
[0017]
本发明还提供了所述的聚酯酰亚胺共聚物的制备方法,包括以下步骤:
[0018]
(1)在惰性气体保护下,以叔丁基对苯二酚双(偏苯三酸酐)、4,4'-(六氟异丙烯)二酞酸酐、芳香二胺单体和芳香酰氯单体为聚合单体,在有机溶剂中进行聚合反应,得到聚酰胺酸溶液;
[0019]
(2)步骤(1)中的聚酰胺酸溶液在脱水剂和催化剂作用下进行酰亚胺化,经后处理得到聚酯酰亚胺共聚物。
[0020]
所述的芳香二胺单体的摩尔量与芳香酰氯单体、叔丁基对苯二酚双(偏苯三酸酐)和4,4'-(六氟异丙烯)二酞酸酐总摩尔量的比例为1:1-1.05。
[0021]
优选的,芳香酰氯单体与叔丁基对苯二酚双(偏苯三酸酐)的摩尔比为0.1-7:1;在上述优选的范围内,随着芳香酰氯含量的增加,聚酯酰亚胺共聚物的热膨胀系数降低,玻璃化转变温度提高。
[0022]
所述的有机溶剂包括n,n-二甲基甲酰胺、n,n-二甲基乙酰胺、环丁砜、二甲基亚砜、n-甲基吡咯烷酮或间甲酚。
[0023]
所述的脱水剂为乙酸酐、丙酸酐、正丁酸酐、戊酸酐和苯甲酸酐中的至少一种;催化剂为吡啶、异喹啉和三乙胺中至少一种;脱水剂与催化剂的摩尔比为1:0.5-1;脱水剂与二胺单体的摩尔比为1-3:1。
[0024]
步骤(2)中,酰亚胺化反应的温度为25-100℃,时间为2-24h。
[0025]
优选的,酰亚胺化反应的温度为70-90℃,时间为2~3h。该反应条件有利于在短时间内实现酰亚胺化完全。
[0026]
本发明还提供了一种聚酯酰亚胺薄膜,由所述的聚酯酰亚胺共聚物制备得到,优选的,将聚酯酰亚胺共聚物溶解得到聚酯酰亚胺溶液,流延或涂布成膜后固化,得到所述的聚酯酰亚胺薄膜。
[0027]
优选的,固化时采用程序升温,程序升温步骤为60-100℃固化1-2小时,100-160℃固化1-2小时,180-250℃固化1-2小时,300-350℃固化1-2小时。
[0028]
进一步优选的,程序升温步骤为80℃固化1-2小时,100℃固化1-2小时,150℃固化1-2小时,200℃固化1-2小时,250℃固化1-2小时。
[0029]
所述的聚酯酰亚胺薄膜的玻璃化转变温度≥300℃,热膨胀系数≤30ppm/k,在400nm的光学透过率≥70%。
[0030]
优选的,所述的聚酯酰亚胺薄膜的玻璃化转变温度为300-350℃,热膨胀系数为10-26ppm/k;在400nm的光学透过率≥74%。
[0031]
与现有技术相比,本发明的有益效果在于:
[0032]
(1)本发明将酰胺结构引入到聚酯酰亚胺体系中,在分子链间形成氢键,增强链的刚性和线性度,从而使得聚酯酰亚胺薄膜的热膨胀系数≤30ppm/k,玻璃化转变温度≥300℃;
[0033]
(2)酰胺基团的存在会相对减少酰亚胺基团的比例,本发明中的聚酯酰亚胺引入强吸电子基团—三氟甲基和大体积侧基—叔丁基,使得产品聚酯酰亚胺薄膜在400nm处的透过率≥70%,在光学领域有着潜在的应用。
附图说明
[0034]
图1为实施例1~4和对比例1中聚酯酰亚胺共聚物的红外光谱图。
具体实施方式
[0035]
下面结合实施例,进一步阐明本发明。应理解,这些实施例仅用于说明本发明,而不用于限制本发明的范围。
[0036]
实施例1
[0037]
(1)氮气保护下,将1.6015g(5.001mmol)2,2'-双(三氟甲基)-4,4'-二氨基联苯,1.3247g(2.575mmol)叔丁基对苯二酚双(偏苯三酸酐)、0.3137g(1.545mmol)对苯二甲酰氯和0.4576g(1.030mmol)4,4'-(六氟异丙烯)二酞酸酐,20.6233g n,n-二甲基乙酰胺加入100ml反应瓶中,室温搅拌10小时,得到透明聚酰胺酸溶液;
[0038]
(2)将透明聚酰胺酸溶液稀释至固含量为10%,加入1.2759g(12.498mmol)乙酸酐以及0.9884g(12.496mmol)吡啶,80℃酰亚胺化反应2.5h,反应结束后冷却至室温,在乙醇/水中沉淀得到纤维状的聚酯酰亚胺共聚物,煮洗烘干,得到聚酯酰亚胺共聚物;
[0039]
(3)将上述聚酯酰亚胺共聚物粉末加入到n,n-二甲基乙酰胺中溶解完全,得到固含量为10%的聚酯酰亚胺溶液,将该溶液涂布到玻璃基板上,置于烘箱中进行升温固化,具体为80℃/2小时、100℃/1小时、150℃/1小时、200℃/1小时、250℃/1小时,降至室温后从烘箱中取出玻璃板,将玻璃板浸泡于水中,聚酯酰亚胺薄膜自动剥离,并将其放入烘箱中干燥备用。
[0040]
本实施例的聚酯酰亚胺共聚物的ft-ir图谱如图1所示,1788,1722,1367cm-1
特征峰的出现能够证明该聚酯酰亚胺共聚物的成功合成。
[0041]
实施例2
[0042]
(1)氮气保护下,将1.6013g(5.000mmol)2,2'-双(三氟甲基)-4,4'-二氨基联苯,1.0597g(2.060mmol)叔丁基对苯二酚双(偏苯三酸酐)、0.4182g(2.060mmol)对苯二甲酰氯和0.4578g(1.030mmol)4,4'-(六氟异丙烯)二酞酸酐,19.7209g n,n-二甲基乙酰胺加入100ml反应瓶中,室温搅拌10小时,得到透明聚酰胺酸溶液;
[0043]
(2)将透明聚酰胺酸溶液稀释至固含量为10%,加入1.2764g(12.503mmol)乙酸酐以及0.9890g(12.503mmol)吡啶,90℃亚胺化2h,反应结束后冷却至室温,在乙醇/水中沉淀得到纤维状的聚酯酰亚胺共聚物,煮洗烘干,得到聚酯酰亚胺共聚物;
[0044]
(3)将上述聚酯酰亚胺共聚物粉末加入到n,n-二甲基乙酰胺中溶解完全,得到固含量为10%的聚酯酰亚胺溶液,将该溶液涂布到玻璃基板上,置于烘箱中进行升温固化,具体为80℃/2小时、100℃/1小时、150℃/1小时、200℃/1小时、250℃/1小时,降至室温后从烘箱中取出玻璃板,将玻璃板浸泡于水中,聚酯酰亚胺薄膜自动剥离,并将其放入烘箱中干燥备用。
[0045]
本实施例的聚酯酰亚胺共聚物的ft-ir图谱如图1所示,1784,1722,1369cm-1
特征峰的出现能够证明该聚酯酰亚胺共聚物的成功合成。
[0046]
实施例3
[0047]
(1)氮气保护下,将1.6012g(4.999mmol)2,2'-双(三氟甲基)-4,4'-二氨基联苯,0.7794g(1.515mmol)叔丁基对苯二酚双(偏苯三酸酐)、0.5127g(2.525mmol)对苯二甲酰氯和0.4487g(1.010mmol)4,4'-(六氟异丙烯)二酞酸酐,29.9219g n,n-二甲基乙酰胺加入100ml反应瓶中,室温搅拌10小时,得到透明聚酰胺酸溶液;
[0048]
(2)将透明聚酰胺酸溶液稀释至固含量为10%,加入1.2762g(12.500mmol)乙酸酐以及0.9885g(12.497mmol)吡啶,80℃酰亚胺化反应2.5h,反应结束后冷却至室温,在乙醇/水中沉淀得到纤维状的聚酯酰亚胺共聚物,煮洗烘干,得到聚酯酰亚胺共聚物;
[0049]
(3)将上述聚酯酰亚胺共聚物粉末加入到n,n-二甲基乙酰胺中溶解完全,得到固含量为10%的聚酯酰亚胺溶液,将该溶液涂布到玻璃基板上,置于烘箱中进行升温固化,具体为80℃/2小时、100℃/1小时、150℃/1小时、200℃/1小时、250℃/1小时,降至室温后从烘箱中取出玻璃板,将玻璃板浸泡于水中,聚酯酰亚胺薄膜自动剥离,并将其放入烘箱中干燥备用。
[0050]
本实施例的聚酯酰亚胺共聚物的ft-ir图谱如图1所示,1786,1726,1369cm-1
特征峰的出现能够证明该聚酯酰亚胺共聚物的成功合成。
[0051]
实施例4
[0052]
(1)氮气保护下,将1.6015g(5.001mmol)2,2'-双(三氟甲基)-4,4'-二氨基联苯,
0.5299g(1.030mmol)叔丁基对苯二酚双(偏苯三酸酐)、0.6273g(3.090mmol)对苯二甲酰氯和0.4576g(1.030mmol)4,4'-(六氟异丙烯)二酞酸酐,28.5461g n,n-二甲基乙酰胺加入100ml反应瓶中,室温搅拌10小时,得到透明聚酰胺酸溶液;
[0053]
(2)将透明聚酰胺酸溶液稀释至固含量为10%,加入1.2766g(12.505mmol)乙酸酐以及0.9890g(12.503mmol)吡啶,70℃酰亚胺化反应3h,反应结束后冷却至室温,在乙醇/水中沉淀得到纤维状的聚酯酰亚胺共聚物,煮洗烘干,得到聚酯酰亚胺共聚物;
[0054]
(3)将上述聚酯酰亚胺共聚物粉末加入到n,n-二甲基乙酰胺中溶解完全,得到固含量为10%的聚酯酰亚胺溶液,将该溶液涂布到玻璃基板上,置于烘箱中进行升温固化,具体为80℃/2小时、100℃/1小时、150℃/1小时、200℃/1小时、250℃/2小时,降至室温后从烘箱中取出玻璃板,将玻璃板浸泡于水中,聚酯酰亚胺薄膜自动剥离,并将其放入烘箱中干燥备用。
[0055]
本实施例的聚酯酰亚胺共聚物的ft-ir图谱如图1所示,1788,1724,1369cm-1
特征峰的出现能够证明该聚酯酰亚胺共聚物的成功合成。
[0056]
实施例5
[0057]
(1)氮气保护下,将1.6812g(5.000mmol)2,2'-双(三氟甲基)-4,4'-二氨基二苯醚,2.0583g(4.001mmol)叔丁基对苯二酚双(偏苯三酸酐)、0.1015g(0.500mmol)间苯二甲酰氯和0.2230g(0.502mmol)4,4'-(六氟异丙烯)二酞酸酐,16.2564g n,n-二甲基乙酰胺加入100ml反应瓶中,室温搅拌10小时,得到透明聚酰胺酸溶液;
[0058]
(2)将透明聚酰胺酸溶液稀释至固含量为10%,加入1.2762g(12.500mmol)乙酸酐以及0.9888g(12.500mmol)吡啶,80℃酰亚胺化反应2.5h,反应结束后冷却至室温,在乙醇/水中沉淀得到纤维状的聚酯酰亚胺共聚物,煮洗烘干,得到聚酯酰亚胺共聚物;
[0059]
(3)将上述聚酯酰亚胺共聚物粉末加入到n,n-二甲基乙酰胺中溶解完全,得到固含量为10%的聚酯酰亚胺溶液,将该溶液涂布到玻璃基板上,置于烘箱中进行升温固化,具体为80℃/2小时、100℃/1小时、150℃/1小时、200℃/1小时、250℃/2小时,降至室温后从烘箱中取出玻璃板,将玻璃板浸泡于水中,聚酯酰亚胺薄膜自动剥离,并将其放入烘箱中干燥备用
[0060]
对比例1
[0061]
(1)氮气保护下,将1.6011g(5.000mmol)2,2'-双(三氟甲基)-4,4'-二氨基联苯,2.0583g(4.001mmol)叔丁基对苯二酚双(偏苯三酸酐)、0.4442g(1.000mmol)4,4'-(六氟异丙烯)二酞酸酐,10.1482g n,n-二甲基乙酰胺加入50ml反应瓶中,室温搅拌10小时,得到透明聚酰胺酸溶液;
[0062]
(2)将透明聚酰胺酸溶液稀释至固含量为10%,加入1.2769g(12.508mmol)乙酸酐以及0.9894g(12.508mmol)吡啶,80℃亚胺化2.5h,反应结束后冷却至室温,在乙醇/水中沉淀得到纤维状的聚酯酰亚胺共聚物,煮洗烘干,得到聚酯酰亚胺共聚物;
[0063]
(3)将上述聚酯酰亚胺共聚物粉末加入到n,n-二甲基乙酰胺中溶解完全,得到固含量为10%的聚酯酰亚胺溶液,将该溶液涂布到玻璃基板上,置于烘箱中进行升温固化,具体为80℃/2小时、100℃/1小时、150℃/1小时、200℃/1小时、250℃/1小时,降至室温后从烘箱中取出玻璃板,将玻璃板浸泡于水中,聚酯酰亚胺薄膜自动剥离,并将其放入烘箱中干燥备用。
[0064]
本对比例的聚酯酰亚胺共聚物的ft-ir图谱如图1所示,1788,1724,1367cm-1
特征峰的出现能够证明该聚酯酰亚胺共聚物的成功合成。
[0065]
样品分析
[0066]
实施例1-4制得的聚酯酰亚胺共聚物的结构式如下式所示,
[0067][0068]
其中,r1为r2为
[0069]
对比例1制得的聚酯酰亚胺共聚物的结构式如下式所示,
[0070][0071]
其中,r1为
[0072]
实施例1-4制得的聚酯酰亚胺薄膜的主要性能如表1所示,相比于对比例,实施例的玻璃化转变温度以及t
400nm
均有所提升,且热膨胀系数有明显的下降。
[0073]
表1实施例1-4所制备的聚酯酰亚胺薄膜的主要性能
[0074][0075]
以上所述的实施例对本发明的技术方案进行了详细说明,应理解的是以上所述的仅为本发明的具体实施例,并不用于限制本发明,凡在本发明的原则范围内所做的任何修改、补充或类似方式替代等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。