1.本发明涉及有机合成技术领域,具体是一种4-羟基-7-甲氧基喹啉的制备方法。
背景技术:
2.4-羟基-7-甲氧基喹啉是用于治疗癌症的蛋白络氨酸激酶(ptk)抑制剂的中间体。现有的合成方法中,环合工艺必须在较高温度(180℃左右,反应2-3h)下进行,生产上要采用油浴加热的方法才能达到反应所需温度,危险系数较高,不易于大规模生产,能耗也相对高,因此需要开发一种低能耗生产上易操作的合成方法。
技术实现要素:
3.本发明的目的是:克服现有技术中的不足,提供一种能耗低,易于大生产的4-羟基-7-甲氧基喹啉的制备方法。
4.为解决上述技术问题,本发明采用的技术方案如下:一种4-羟基-7-甲氧基喹啉的制备方法,所述制备方法包括以下步骤:s1 在反应步骤中加入乙醇、原甲酸三乙酯、丙二酸环亚异丙酯,升温,滴加3-甲氧基苯胺,滴加完毕继续回流,hplc跟踪反应,反应结束后降温至室温,离心分离,用冰乙醇洗涤,干燥得到中间体2,2-二甲基-5-[(3-甲氧基苯基氨基)亚甲基]-1,3-二氧六环-4,6-二酮,滤液回收乙醇套用;s2 在反应容器中加入氯苯、步骤s1中制得 的中间体和催化剂,搅拌加热反应一段时间,hplc跟踪反应,反应结束后冷却至室温,离心得到粗产物;s3 提纯干燥后得到产物。
[0005]
进一步的,所述步骤s1中升温至75~85℃,3-甲氧基苯胺滴加结束后继续回流2~3h。
[0006]
进一步的,所述步骤s1中乙醇、原甲酸三乙酯、丙二酸环亚异丙酯和3-甲氧基苯胺的质量比为(2.5~2.67):(2.5~2.67):(1.2~1.26):1。
[0007]
进一步的,所述步骤s1中干燥温度为55~60℃。
[0008]
进一步的,所述步骤s2中氯苯与中间体和催化剂的质量比为(2.61~2.75):1:(0.05~0.065)。
[0009]
进一步的,所述步骤s2中的催化剂选用路易斯酸。
[0010]
进一步的,所述催化剂选氯化锌、氯化铝、氯化锡或对甲苯磺酸中的一种。
[0011]
进一步的,所述步骤s2中反应温度为120~130℃,搅拌时间为2~3小时。
[0012]
进一步的,所述步骤s3包括以下步骤:1)用水在20~30℃下搅拌粗产物2~3小时,然后离心分离;
2)步骤1)中得到的固体用乙醇重结晶,降温至10~20℃搅拌1~2h,离心分离,固体用冰乙醇洗涤;3)步骤2)中得到的固体在真空中干燥,得到淡黄色固体化合物。
[0013]
进一步的,所述步骤1)中水和粗品的比例为1:1~1.5:1,步骤2)中乙醇和粗品的比例为1.5:1~2:1,步骤3)中的干燥温度为70~80℃。
[0014]
采用本发明的技术方案的有益效果是:相比于现有的合成方法中,环合工艺必须在较高温度180℃左右反应2~3h,本发明第二步环合反应中加入了催化剂并将溶剂换成氯苯,降低了反应温度(即120~130℃),从而只需要水蒸气加热就可以达到反应所需温度,更易于大生产,安全系数高,也节省了能耗。
附图说明
[0015]
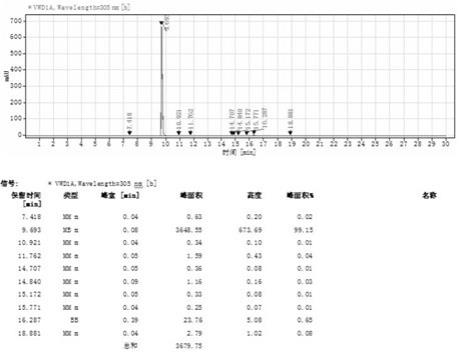
图1为本发明中最终产品的hplc图谱;图2为本发明中最终产品的h1nmr图谱。
具体实施方式
[0016]
下面将结合本发明具体实施例对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0017]
本发明中的是室温是指20-30℃。
[0018]
图1为本发明中最终产品的hplc图谱。
[0019]
图2为本发明中最终产品的h1nmr图谱。
[0020]
实施例1(1)合成2,2-二甲基-5-[(3-甲氧基苯基氨基)亚甲基]-1,3-二氧六环-4,6-二酮在反应容器中加入乙醇400kg、原甲酸三乙酯400kg、丙二酸环亚异丙酯180kg,升温,滴加3-甲氧基苯胺150kg,滴加完毕,再回流2h,hplc跟踪反应,待原料含量《0.5%,停止反应,降温至室温20度,离心,用冰乙醇洗涤,55-60℃干燥得到中间体(304kg,收率89.7%)。
[0021]
(2)合成4-羟基-7-甲氧基喹啉在反应容器中加入氯苯550kg,2,2-二甲基-5-[(3-甲氧基苯基氨基)亚甲基]-1,3-二氧六环-4,6-二酮 200kg,催化剂氯化锌10kg,将反应物加热至130℃并继续搅拌2小时;hplc跟踪反应,待原料含量《0.5%,停止反应,冷却到室温,离心得到黄色固体的粗产物130kg;用150kg水在20-30℃下搅拌粗产物2小时,然后离心,得到的固体135kg,将其加入到反应容器中,加入250kg乙醇升温至溶解,降温至10℃搅拌1h,离心,用冰乙醇洗涤,在真空中干燥,得到淡黄色固体(115kg,收率91.3%)。
[0022]
实施例2
(1)合成2,2-二甲基-5-[(3-甲氧基苯基氨基)亚甲基]-1,3-二氧六环-4,6-二酮在反应容器中加入乙醇450kg、原甲酸三乙酯450kg、丙二酸环亚异丙酯220kg,升温,滴加3-甲氧基苯胺175kg,滴加完毕,再回流2h,hplc跟踪反应,待原料含量《0.5%,停止反应,降温至室温30度,离心,用冰乙醇洗涤,55-60℃干燥得到中间体(352kg,收率89.0%)。
[0023]
(2)合成4-羟基-7-甲氧基喹啉在反应容器中加入氯苯600kg,2,2-二甲基-5-[(3-甲氧基苯基氨基)亚甲基]-1,3-二氧六环-4,6-二酮 220kg,催化剂氯化锌14kg,将反应物加热至120℃并继续搅拌3小时;hplc跟踪反应,待原料含量《0.5%,停止反应,冷却到室温,离心得到黄色固体的粗产物145kg;用180kg水在20-30℃下搅拌粗产物2小时,然后离心,得到的固体155kg,将其加入到反应容器中,加入290kg乙醇升温至溶解,降温至15℃搅拌2h,离心,用冰乙醇洗涤,得到的固体在真空中干燥,得到淡黄色固体(126kg,收率91.0%)。
[0024]
实施例3(1)合成2,2-二甲基-5-[(3-甲氧基苯基氨基)亚甲基]-1,3-二氧六环-4,6-二酮在反应容器中加入乙醇500kg、原甲酸三乙酯500kg、丙二酸环亚异丙酯250kg,升温,滴加3-甲氧基苯胺200kg,滴加完毕,再回流2h,hplc跟踪反应,待原料含量《0.5%,停止反应,降温至室温30度,离心,用冰乙醇洗涤,55-60℃干燥得到中间体(408kg,收率90.3%)。
[0025]
(2)合成4-羟基-7-甲氧基喹啉在反应容器中加入氯苯600kg,2,2-二甲基-5-[(3-甲氧基苯基氨基)亚甲基]-1,3-二氧六环-4,6-二酮 230kg,催化剂氯化锌15kg,将反应物加热至125℃并继续搅拌3小时;hplc跟踪反应,待原料含量《0.5%,停止反应,冷却到室温,离心得到黄色固体的粗产物155kg;用200kg水在25℃下搅拌粗产物2小时,然后离心,得到的固体170kg,将其加入到反应容器中,加入310kg乙醇升温至溶解,降温至20℃搅拌2h,离心,用冰乙醇洗涤,得到的固体在真空中干燥,得到淡黄色固体(131kg,收率90.5%)。
[0026]
此外,应当理解,虽然本说明书按照实施方式加以描述,但并非每个实施方式仅包含一个独立的权利方案,说明书的这种叙述方式仅仅是为清楚起见,本领域技术人员应当将说明书作为一个整体,各实施例中的技术方案也可以经适当组合,形成本领域技术人员可以理解的其他实施方式。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。