基于alg包裹的具有活性氧响应的apd-l1前药水凝胶及其制备方法
技术领域
1.本发明属于水凝胶纳米复合材料制备及其应用领域,特别涉及一种基于alg(海藻酸钠)包裹的具有活性氧响应的apd-l1前药水凝胶及其制备方法。
背景技术:
2.近年来,关于癌症治疗的方法被很多人报道,传统的放射治疗和化疗仍是现在主要的治疗手段,但由放疗和化疗引起的耐药性和副作用是不可忽视的问题。
3.免疫治疗在癌症治疗中显示了巨大的前景,不仅是2013年《科学》杂志评选的年度最重要的科学突破,2018年的诺贝尔生理学或医学奖也发给了相关研究。肿瘤免疫治疗主要包括免疫检查点抑制剂、细胞治疗、肿瘤疫苗等。但在临床应用中,只有一小部分晚期癌症患者对免疫检查点抑制剂单药治疗有反应,且现有方法对免疫调动不足,或者通过自身肿瘤微环境去促进免疫因子的释放不足。
技术实现要素:
4.本发明的目的是克服现有材料对免疫调动不足,或者通过自身肿瘤微环境去促进免疫因子的释放不足等问题,提供一种基于alg包裹的具有活性氧响应的apd-l1前药水凝胶制备与应用。该方法制备得到的水凝胶纳米平台具备优异的生物相容性和稳定性,具有良好的杀伤肿瘤和免疫治疗效果,为瘤内给药和免疫应激一体化平台的开发提供了一种新方法,应用前景广阔。
5.为了达到上述目的,本发明提供了一种基于alg包裹的具有活性氧响应的apd-l1前药水凝胶的制备方法,包含:
6.步骤1,制备pf纳米颗粒:
7.步骤1.1,以 3价铁盐、 2价铁盐、bsa为原料,水浴加热,碱性条件下搅拌反应,获得bsa包裹fe3o4纳米颗粒,记为bsa@fe3o4;
8.步骤1.2,将ppix、edc
·
hcl、n-羟基丁二酰亚胺nhs混合溶解后,加入bsa@fe3o4,搅拌反应,将ppix负载到bsa@fe3o4上,得到ppix-bsa@fe3o4,记为pf纳米颗粒;
9.步骤2,制备apd-l1前药:以活性氧响应性的连接物、bsa、pd-l1抗体为原料,混合反应,得到apd-l1前药;
10.步骤3,将海藻酸钠alg、pf纳米颗粒、apd-l1前药充分混合,得到alg@ppix-bsa@fe3o4&apd-l1,将其加入到含有ca
2
的溶液中,成胶,得到基于alg包裹的具有活性氧响应的apd-l1前药水凝胶。
11.可选地,步骤1.1中,碱性条件是指加入氢氧化钠、氢氧化钾、氨水中的任意一种或任意两种以上的混合。
12.可选地,步骤1.1中,所述 3价铁盐为fecl3·
6h2o,所述 2价铁盐为fecl2·
4h2o,碱性条件是指加入naoh,其中,fecl3·
6h2o,fecl2·
4h2o,naoh的摩尔比为1:1:100~1:1:
500。
13.可选地,步骤1.2中,ppix、edc
·
hcl、nhs的摩尔比为1:3:3~1:10:10。
14.可选地,所述pf纳米颗粒中,ppix在bsa@fe3o4的表面负载率为59.4%。
15.可选地,步骤2中,先将活性氧响应性的连接物、edc
·
hcl、nhs在dmso中混合以对活性氧响应性的连接物进行活化处理,将bsa、pd-l1抗体在ph为7.4的pbs中混合,搅拌状态下将活化的活性氧响应性的连接物加入到bsa与pd-l1抗体的混合液中,在4℃、黑暗条件下反应8h~16h,将得到的产物在4℃下透析得到apd-l1前药纳米颗粒。
16.可选地,活性氧响应性的连接物、edc
·
hcl、nhs的摩尔比为1:3:3~1:10:10,bsa与pdl1抗体的摩尔比为1:1~5:1,dmso与ph7.4的pbs的体积比例为1:20~1:100。
17.可选地,透析条件为:用截留分子量为8000~14000的透析袋透析2-3天。
18.可选地,步骤3中,alg的最终浓度为5mg/ml。
19.本发明还提供了一种采用上述制备方法得到的基于alg包裹的具有活性氧响应的apd-l1前药水凝胶,其结构式为alg@ppix-bsa@fe3o4&apd-l1,包含:海藻酸钠载体alg、bsa、fe3o4纳米颗粒、原卟啉ppix、apd-l1前药,其中,bsa包裹fe3o4纳米颗粒并外接ppix形成ppix-bsa@fe3o4颗粒,其与apd-l1前药均匀分散在海藻酸钠载体中,与ca
2
交换得到所述的水凝胶。
20.本发明的有益效果:
21.(1)本发明制备得到的appf前药水凝胶,内部的pf颗粒分布均一,尺寸和粒子形貌均可控,apd-l1前药纳米颗粒也具有良好的分散性和稳定性,克服了现在前药水凝胶的一些缺陷。
22.(2)本发明采用bsa包覆以稳定fe3o4纳米颗粒,再通过ppix的羧基与bsa的氨基反应形成的酰胺键将光敏剂ppix外接在bsa包裹的fe3o4表面形成pf纳米颗粒,增强了ppix和fe3o4的稳定性和生物相容性,也增加了pf纳米颗粒的功能性,使得pf纳米颗粒具有pdt(光动力学治疗)和cdt(化学动力学)双功能。
23.光动力疗法和化学动力疗法均是通过产生有毒性的活性氧(1o2和
·
oh)达到杀死肿瘤的目的。与传统治疗手段相比,光动力疗法有着无创性、低毒性且不引起耐药性的天然优势,化学动力疗法是通过fe、mn、cu等材料与肿瘤内部过高的h2o2反应在肿瘤内部产生有毒性的
·
oh达到杀伤肿瘤的效果,其具有无毒稳定且制备简单的优点。
24.(3)本发明的apd-l1前药纳米颗粒,通过用活性氧响应性的连接物(提供羧基)将pd-l1抗体(提供氨基)键合连接在一起,再通过bsa包裹(提高生物相容性),形成具有活性氧响应性的apd-l1纳米颗粒(apd-l1前药),具有可控释放的性能。ppix通过660nm的近红外光照射后产生的单线态氧和fe3o4遇到肿瘤内部的过氧化氢反应产生的羟基自由基都属于活性氧,本发明通过给予外部红外光刺激和肿瘤内部过氧化氢的反应瞬间提高肿瘤内部的活性氧含量来打断活性氧响应性的连接物,使得apd-l1前药纳米颗粒由一个大的纳米颗粒变成单个的pd-l1抗体,即实现了pd-l1抗体的可控释放。
25.(4)本发明制备的appf纳米平台在具有可控pd-l1抗体释放的能力同时,还具有pdt,cdt以及免疫治疗的联合治疗策略,是一种有潜力的可实现pdt,cdt协同免疫治疗的多功能纳米平台,具有很好的市场应用前景。
附图说明
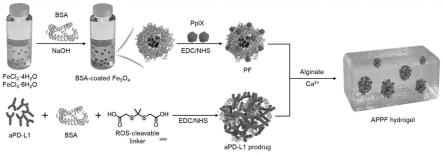
26.图1为本发明的制备appf前药水凝胶平台的反应过程图。
27.图2为本发明的bsa包裹fe3o4和pf的紫外光谱图(图中的a)和荧光谱图(图中的b)。
28.图3为本发明的apd-l1前药纳米颗粒的sem图。
29.图4为本发明的bsa包裹fe3o4,pf,apd-l1前药的粒径分布(图中的a)与表面电势图(图中的b)。
30.图5为本发明的appf前药水凝胶在形成水凝胶过程中的实物照片。
31.图6为本发明中的apf与appf在形成水凝胶之后的sem图片,其中,a代表apf,b代表appf。
32.图7为本发明的apf、appf前药水凝胶在形成水凝胶之后的tmb实验,其中,a代表apf在形成水凝胶前的tmb实验,b代表appf在形成水凝胶前的tmb实验。
33.图8为本发明的pf、apf、appf在溶液和凝胶状态下评估的单线态氧生成能力。
34.图9为本发明的appf前药水凝胶在不同条件下(100μm的h2o2,660nm激光照射5min以及100μm的h2o2和660nm激光照射5min)pd-l1抗体的释放曲线示意图。
35.图10为本发明的appf前药水凝胶、对照组apf水凝胶分别与4t1细胞共孵育24h后的细胞毒性实验结果。
36.图11为本发明的appf前药水凝胶、对照组apf水凝胶在不同处理条件下与4t1细胞共孵育24h后的细胞毒性实验结果。
37.图12为本发明的appf前药水凝胶通过瘤内注射入小鼠肿瘤部位,利用660nm激光照射5min后,记录24天内小鼠原发肿瘤体积(a),24天内小鼠转移肿瘤体积(b),小鼠在50天的存活率(c)以及在治疗过程中的体重变化(d)。
具体实施方式
38.目前的免疫治疗研究,大多数都是在于递送系统,以解决药物递送的靶向性、跨越生物屏障及降低毒副作用等问题。而大部分的纳米药物是基于纳米载体技术实现靶向性递送,比较依赖于epr效应(高通透性和滞留效应,enhanced permeability and retention effect)。该效应在动物模型上比较显著,但在人类肿瘤中的epr效应是否显著目前存在很大争议,因此,瘤内给药就显得更有价值。
39.海藻酸钠是一种天然多糖,具有药物制剂辅料所需的稳定性、溶解性、粘性和安全性。已在生物材料和医药领域得到了广泛应用。liu等人(advanced materials,2021,33,2007910)制备了一个具有藻酸盐与三磷酸腺苷(atp)响应的水凝胶,可在达到在注射奥沙利珀时引起atp的释放导致免疫佐剂cpg的释放,然而该智能水凝胶是依靠的肿瘤内部自身产生的atp去促进药物释放;其调动免疫不足是因为其通过凋亡细胞释放的atp去响应性释放接在水凝胶上面的免疫药物cpg。li等人(j.mater.chem.b.2021.9.5255-5263)制备了可以抑制转移具有联合治疗的水凝胶,通过用alg包裹dna酶、二氧化锰和ppix,在局部对肿瘤细胞的治疗有很好的效果,然而该水凝胶可能在免疫上面的调动不足,不能起到通过局部治疗达到全身免疫的效果。
40.本发明通过将其他疗法与pd-l1抗体免疫治疗相结合,提供一种基于alg包裹的具有活性氧响应的apd-l1前药,其内含的ppix、fe3o4促进产生的活性氧可使肿瘤从冷肿瘤变
为热肿瘤,从而提高免疫系统对肿瘤细胞的识别能力(提高免疫细胞的数量和战斗力),通过激光照射的外部刺激和肿瘤内部较高过氧化氢微环境这种内外结合响应的方式去促进pd-l1抗体释放,进而可以达到通过对局部的肿瘤进行治疗去促进全身免疫。
41.对肿瘤微环境中内部刺激(如ph值、gsh及氧化还原电位)敏感的可切割连接物或特定肽,已被广泛用于构建前药。因此,通过对纳米平台的合理设计,开发一种响应性的新型、高效纳米平台成为可能。
42.检索国内外有关纳米递药平台方面的文献和专利结果表明:目前还没有发现基于alg包裹的具有活性氧响应的pd-l1抗体前药来实现光动力疗法和化学动力疗法协同促进免疫治疗的多功能纳米颗粒的制备与应用方面的报道。
43.为了提高免疫治疗的效果和患者反应率,本发明将多种治疗方法结合去进行治疗。在响应性反应后转化为生物活性形式的前药,可以大大降低药物脱靶毒性。
44.本发明利用bsa包裹fe3o4纳米颗粒外接ppix,利用bsa包裹活性氧响应的连接物键合连接的pd-l1抗体,然后用alg包裹之后形成原位水凝胶,实现了pdt,cdt和免疫疗法三者的协同治疗从而制备了具有优异的水凝胶纳米平台,满足了通过局部治疗去调动全身免疫系统的需求。水凝胶的制备方法具体包含:
45.步骤1,制备pf纳米颗粒:
46.步骤1.1,以 3价铁盐、 2价铁盐、bsa为原料,水浴加热,碱性条件下搅拌反应,获得bsa包裹fe3o4纳米颗粒,记为bsa@fe3o4;所述碱性条件是指加入氢氧化钠、氢氧化钾、氨水中的任意一种或任意两种以上的混合。所述 3价铁盐为fecl3·
6h2o,所述 2价铁盐为fecl2·
4h2o,碱性条件是指加入naoh,其中,fecl3·
6h2o,fecl2·
4h2o,naoh的摩尔比可为1:1:100~1:1:500。
47.步骤1.2,将ppix、edc
·
hcl、n-羟基丁二酰亚胺nhs混合溶解后,加入bsa@fe3o4,搅拌反应,将ppix负载到bsa@fe3o4上,得到ppix-bsa@fe3o4,记为pf纳米颗粒;其中,ppix、edc
·
hcl、nhs的摩尔比可为1:3:3~1:10:10。
48.步骤2,制备apd-l1前药:以活性氧响应性的连接物、bsa、pd-l1抗体为原料,混合反应,得到apd-l1前药;一些实施例中,先将活性氧响应性的连接物、edc
·
hcl、nhs在dmso中混合,其中,活性氧响应性的连接物、edc
·
hcl、nhs的摩尔比可为1:3:3~1:10:10,以对活性氧响应性的连接物进行活化处理,将bsa、pd-l1抗体在ph为7.4的pbs中混合,其中,bsa与pdl1抗体的摩尔比可为1:1~5:1,dmso与ph7.4的pbs的体积比例为1:20~1:100,搅拌状态下将活化的活性氧响应性的连接物加入到bsa与pd-l1抗体的混合液中,在4℃、黑暗条件下反应8h~16h,将得到的产物在4℃下透析得到apd-l1前药纳米颗粒。其中,透析条件为:用截留分子量为8000~14000的透析袋透析2-3天。
49.步骤3,将海藻酸钠alg、pf纳米颗粒、apd-l1前药充分混合,其中,alg的最终浓度为5mg/ml,得到alg@ppix-bsa@fe3o4&apd-l1,将其加入到含有ca
2
的溶液中,成胶,得到基于alg包裹的具有活性氧响应的apd-l1前药水凝胶。
50.下面将结合附图和实施例对本发明的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
51.本发明的实施例中所用到的主要材料和试剂来源如表1所示:
52.表1所用到的主要材料和试剂
[0053][0054]
本发明的实施例使用扫描电子显微镜(sem),电势粒径,紫外-可见分光光度计(uv-vis),荧光分光光度计,细胞活力分析(cck-8测试)以及体外、体内联合治疗表征本发明制备的具有响应性的alg包裹的pd-l1抗体水凝胶纳米平台。实施例1
[0055]
制备appf前药水凝胶平台的具体过程如图1所示:
[0056]
(1)先向烧瓶中加入157mg的fecl3·
6h2o和89mg的fecl2·
4h2o,再加入8ml的水使其在超声状态下完全溶解。
[0057]
(2)称取1g的naoh粉末和250mg的牛血清白蛋白(bsa),分别加入1ml的水在超声的情况下使其完全溶解。本步骤中,naoh也可以用氨水或氢氧化钾替换。
[0058]
(3)将(1)中的溶液在水浴锅中加热到80℃,再将(2)中的naoh和bsa溶液在搅拌的情况下加入(1)中,持续反应半小时。反应完毕后,待到水浴锅温度降到30℃以下时,将产物放在磁力搅拌器上,通过用磁力对bsa包裹fe3o4进行沉降与吸走上清液的方法除去未反应的反应物与naoh,沉降的次数5次为宜,沉降过后即可得到需要的bsa包裹fe3o4纳米颗粒bsa@fe3o4。由图4可以看出bsa包裹fe3o4纳米颗粒的尺寸为59.1nm(见图中的a),表面电势为-17.1mv(见图中的b)。
[0059]
(4)将2mg的原卟啉ix(ppix),7mg的1-乙基-(3-二甲基氨基丙基)碳二亚胺盐酸盐
(edc
·
hcl)与10mg的n-羟基丁二酰亚胺(nhs)溶解于n,n-二甲基甲酰胺(dmf)中,在黑暗条件下剧烈搅拌2小时,得到ppix溶液。随后将ppix溶液加入到1.6ml的bsa包裹fe3o4纳米颗粒溶液中(fe的浓度为5mg/ml),继续在黑暗条件下反应8h,室温条件下将得到的产物在去离子水中透析两天(透析袋尺寸:mw=14000d),冻干之后可以得到ppix负载在bsa包裹fe3o4上的纳米颗粒ppix-bsa@fe3o4,记为pf纳米颗粒。如图2的a所示,在500-700nm的三段等离子偏振峰可以看出ppix存在于pf的体系中,并可计算出ppix在bsa包裹fe3o4上的负载率为59.4%。其中,负载率的计算方法为:先采用等离子发射体光谱仪将bsa包裹fe3o4和pf纳米颗粒中铁的浓度定为一样,然后再测试bsa包裹fe3o4和pf纳米颗粒的紫外谱图,通过紫外谱图(图2的a)上的642nm吸光度的差值即可算出ppix的实际含量,再与投料量作对比,即可算出ppix的负载率,ppix的负载量公式为:[abs(pf
642nm-bsa包裹fe3o
4 642nm
)-0.02281]/0.02393。如图2的b可见,引入ppix之后荧光信号明显增强,这也可以证明ppix的成功引入。根据图4可以看出pf的尺寸为64nm(见图中的a),表面电势为-17.3mv(见图中的b)。
[0060]
(5)先将2mg的活性氧响应性的连接物(合成方法见文献angew.chem.int.ed.2019,58,12680-12687),10mg的edc
·
hcl与12mg的nhs在100ul的dmso中混合,混合之后在200rpm/min的摇床上反应两小时,进行活化。再将2mg的bsa,1mg的pd-l1抗体在ph为7.4的磷酸盐缓冲液(pbs)中溶解并混合均匀。将上述活化后的活性氧响应性的连接物的溶液缓慢加入到bsa与pd-l1抗体的混合液中,其中混合液在加入的过程中处于搅拌的状态,在4℃黑暗条件下反应12h,将得到的产物在4℃下透析两天即得到apd-l1前药纳米颗粒。如图3所示,apd-l1纳米颗粒显示近似球状,通过标尺可以看出apd-l1前药纳米颗粒在250nm左右。图4(a)的结果可以看出apd-l1前药的粒径为257.8nm(见图中的a),表面电势为-25.3mv(见图中的b)。
[0061]
本发明的纳米颗粒bsa包裹fe3o4,pf,apd-l1前药纳米颗粒的表面电势和粒径如图4所示,可见,bsa包裹fe3o4和pf的表面电势没有太大差异,都为-17mv左右。这证明了ppix的负载不会改变bsa包裹带来的增强生物相容性效果,apd-l1前药表现出更低一些的电位,为-25.3mv。
[0062]
(6)将10mg/ml的海藻酸钠(alg)、500ug/ml的pf与水按照体积比5:4:1的比例充分混合之后即得到了对照组alg@ppix-bsa@fe3o4(记为apf)。
[0063]
将10mg/ml的alg与500ug/ml的pf,1000ug/ml的apd-l1前药按照体积比5:4:1的比例充分混合之后即得到了alg@ppix-bsa@fe3o4&apd-l1前药水凝胶,即具有活性氧响应的apd-l1前药水凝胶,记为appf。
[0064]
对appf成胶过程的照片与成胶之后的sem进行分析,实验的操作方法如下:将500μl的appf溶液注入2ml的注射器内,然后缓慢的将appf溶液推入1.8mm的ca
2
溶液中,在此过程中用摄影机记录成胶的过程,并在成胶2h后分别用镊子和扫描电镜(sem)测试形成的水凝胶的成胶性能。如图5所示,水凝胶在ca
2
溶液中可以顺利的由溶液转换为凝胶状态,并在形成凝胶两个小时后还有一个很好的凝胶状态。如图6所示,在sem图中可以清晰的得出,在alg与ca
2
相遇时可以顺利形成水凝胶,本实施例所构建的appf前药水凝胶表面有很多的纳米颗粒,这也证明了该水凝胶具有好的包裹性能。
[0065]
羟基自由基(
·
oh)测试
[0066]
本发明制备的appf前药水凝胶在凝胶状态下通过于tmb和100μm的h2o2反应,去检
测
·
oh的产生。具体如下:
[0067]
取实施例1中(6)所得的apf与appf,分别用1.6mm的3,3
′
,5,5
′‑
四甲基联苯胺(tmb)溶液配制成fe浓度为20ug/ml的材料,分别装在4ml的微型离心管中,各2管。再向两组中其中一管加入100μm的h2o2,并设置h2o2加tmb溶液作为对照组。待反应30min后用紫外分光光度计检测tmb在600-700nm处的特征峰,如图7所示,其中,a代表apf对照组,b代表本发明的appf实验组。实验结果显示:在30min之内,apf和appf水凝胶在600-700nm处都有很强的tmb的紫外吸收峰,证明前药水凝胶appf就可以产生大量的
·
oh,有着优异的化学动力学治疗潜力。
[0068]
单线态氧(1o2)测试
[0069]
对含有ppix的纳米颗粒pf,apf,appf采用660nm激光器照射的方式,再用单线态氧荧光探针(sosg)对激光照射过程中的单线态氧进行荧光检测,具体如下:
[0070]
分别取实施例1中(6)所得的apf与appf,分别采用超纯水和1.8mm的ca
2
溶液配制成ppix浓度为5μg/ml的溶液和凝胶。再将150μm的单线态氧荧光探针(sosg)溶液与含有纳米颗粒或水凝胶的溶液混合。混合完成后,使用荧光光谱仪测量溶液在528nm处的荧光强度基线(f0),发射光谱为488nm,激励为500-700nm。然后用功率密度为1w/cm2的660nm激光照射溶液。照射不同时间后,测定溶液在528nm处的荧光强度(f)。计算f/f0比值,验证1o2的生成。实验结果如图8所示:在照射10min后,pf的溶液和apf的水凝胶体系都可以使ppix产生1o2的能力增加1.8倍,而appf因为活性氧响应性的连接物需要消耗部分ros打断所以只上升到1.7倍。但这个结果可以很好的说明了不论凝胶和溶液,都具有很好的光动力治疗潜力。
[0071]
活性氧促进pd-l1抗体的释放测试
[0072]
将pd-l1抗体用fitc进行荧光标记,通过测定fitc的荧光强度去确定pd-l1抗体的释放。
[0073]
取实施例1中(6)所得的appf前药溶液4ml,按照一个瓶子1ml的分配方法将appf前药溶液分别注射到4个含9ml的1.8mm ca
2
溶液的瓶子中,在appf溶液顺利形成水凝胶之后,分别对凝胶进行不同处理:660nm激光照射(laser组)10min;加入100μm的h2o2;660nm激光器照射10min并加入100μm的h2o2;不做任何处理appf凝胶作为对照组(control)。然后分别在处理之后的0h、1h、2h、4h、6h、12h和24h收集凝胶外部3ml的ca
2
溶液,并向凝胶溶液中加入3ml的1.8mm的ca
2
溶液(由于在前一步已经收集了外部的溶液3ml,需要保持总体积不变)。收集到的溶液用以检测体系中pd-l1抗体的释放量。pd-l1抗体的释放曲线如图9所示:在660nm激光和h2o2双重刺激分别产生的oh
·
和1o2的双重作用下,24h之后pd-l1抗体的释放量达到69.9%,比只有660nm激光(35.1%)和只有h2o2(29%)都要高,取得了意料不到的协同效果。这证明了本发明的前药水凝胶系统对于pd-l1抗体有很好的控制释放能力,ppix和fe3o4所产生的1o2和
·
oh在促进apd-l1前药纳米颗粒释放时有很好的协同作用。
[0074]
cck-8细胞毒性测试
[0075]
用4t1细胞研究本发明制备得到的纳米材料的细胞相容性,具体如下:
[0076]
取实施例1中(6)的apf和appf分别用培养基配置成fe浓度为200μg/ml的母液,之后梯度稀释为100、50、25、15μg/ml的材料。取培养好的4t1细胞种于96孔板中,按照1万细胞/孔的密度接种,每孔体积100μl。培养24h后,细胞密度为50%左右,用100μl的pbs清洗2-3次,之后加入上述各稀释梯度的材料,与细胞共培养24h。每个梯度做5个平行孔,以空白培
养基作为空白对照。再孵育24h后用100μl pbs清洗3次,之后每孔加90μl无血清培养基和10μl cck8溶液,在37℃的条件下孵化2h,用酶标仪检测450nm处吸光度值。cck-8法检测细胞活力结果表明,在0-200μg/ml范围内,apf和appf的细胞存活率都在80%以上,这证明我们的材料没有明显的细胞毒性,也表明该水凝胶有良好的细胞相容性(图10)。
[0077]
cck-8治疗效果测试
[0078]
用4t1细胞研究本发明制备得到的纳米材料(apf和appf)的细胞治疗效果,具体如下:
[0079]
取实施例1中(6)的apf和appf,分别用培养基配置成fe浓度为200μg/ml的母液。取培养好的4t1细胞种于96孔板中,按照1万细胞/孔的密度接种,每孔体积100μl。培养24h后,细胞密度为50%左右,用100μl的pbs清洗2-3次,随后各个孔加入apf和appf的溶液,以空白培养基作为对照组。孵育12h后对细胞做不同处理,加100μm的h2o2,660nm激光(laser)照射,660nm激光和100μm的h2o2共同作用。处理之后再与细胞共培养12h后用100μl pbs清洗3次,之后每孔加90μl无血清培养基和10μl cck8溶液,在37℃的条件下孵化2h,用酶标仪检测450nm处吸光度值。如图11所示,cck-8法检测细胞活力结果表明,apf和appf在经过660nm激光和h2o2处理双重刺激后,对4t1细胞具有明显的杀伤作用,即,对4t1的细胞活力有很好的抑制作用,这也预示其有很好的体内联合治疗效果。
[0080]
体内联合治疗测试
[0081]
将appf水凝胶的溶液瘤内注射到有着50mm3大小的4t1荷瘤小鼠内部,appf在小鼠肿瘤内部原位形成水凝胶,利用近红外激光(660nm 1w cm-2
)对肿瘤照射10min后,在第0天到24天记录原发瘤的大小(a),转移瘤的大小(b),小鼠存活率(c)和老鼠体重(d),参见图12。具体操作如下:
[0082]
将实施例1中(6)所得产品(apf和appf),分别用无菌pbs缓冲液配制成fe浓度为200μg/ml的apf和appf溶液。取20μl通过瘤内注射在体重为20g的4t1荷瘤小鼠肿瘤内,2h后利用660nm的激光照射小鼠肿瘤部位(照射时间为10min,照射功率为1w/cm2)。之后记录24天内小鼠肿瘤体积、小鼠体重和小鼠存活率。以瘤内注射pbs(无激光照射),free a pd-l1,apf(无激光照射),appf颗粒(无激光照射)作为空白对照,apf纳米颗粒(660nm激光照射)。小鼠肿瘤部位的治疗测试结果显示:appf前药水凝胶具有优异的光动力和化学动力治疗相结合效果,能在近红外激光照射下通过提高局部的ros浓度杀死肿瘤细胞,也能通过pd-l1抗体引起的免疫检查点阻断治疗肿瘤,从而使小鼠的肿瘤消失并得到痊愈(图12的a),而且接受了appf前药水凝胶治疗的老鼠在接种肿瘤50天之后还有着100%的存活率(图12的c),这是通过调控pd-l1抗体释放而对老鼠全身免疫系统的积极响应(图12的b),这证明本方法合成的appf具有光动力和化学动力学治疗相协同免疫治疗的功能。原发瘤是通过appf水凝胶治疗的,转移瘤是没有经过水凝胶处理的,就是通过缓释抗体获得持续的免疫激活,激活的肿瘤特异性t细胞可以迁移到全身各个系统,并协同抗原暴露实现全身性肿瘤杀伤。同时我们还对肿瘤的体重进行了长达24天的监测,从图12的d可以看出经过凝胶处理的老鼠体重与未经处理的对照组体重之间差别不大,这证明我们的水凝胶系统对老鼠没有毒性,是一种很友好的材料。
[0083]
上述小鼠肿瘤部位的治疗效果表明appf水凝胶有着很好的产生活性氧的能力,在肿瘤内部的过氧化氢和外部激光的双重作用下通过在肿瘤部位产生
·
oh和1o2,一方面使
pdl1抗体释放,一方面通过光动力治疗和化学动力治疗去杀死肿瘤细胞,从而实现了对肿瘤的彻底消融。
[0084]
免疫治疗对转移瘤的抑制结果
[0085]
在小鼠上建立远端瘤研究了由pdt和cdt介导的免疫原性死亡(icd)和pdl1抗体介导的免疫检查点抑制(icb)通过调动小鼠的免疫系统对于肿瘤的治疗效果。研究了远端肿瘤在24天的生长情况,由图12的b可以初步看出,单一的治疗手段,比如free apd-l1组的icb和apf laser组的icd治疗手段对远端肿瘤都没有很好的抑制效果,而appf laser组的远端肿瘤在icd和icb的双重作用下,可以在第14天的时候完全将肿瘤消除,而且在第24天时也没有复发,这可以初步证明老鼠是通过激活的老鼠免疫功能去消除远端肿瘤。
[0086]
综上所述,本发明采用bsa稳定fe3o4纳米颗粒,再通过酰胺键将光敏剂ppix外接在bsa包裹的fe3o4表面形成pf纳米颗粒,增强了ppix和fe3o4的稳定性和生物相容性,并使得pf纳米颗粒具有pdt(光动力学治疗)和cdt(化学动力学)双功能,且二者协同增强。通过用ros响应性清除剂将pd-l1抗体连接在一起,使得apd-l1形成具有ros响应性的纳米颗粒,具有可控释放的性能。本发明形成的基于alg包裹的具有活性氧响应的apd-l1前药水凝胶,光动力学、化学动力学治疗协同增效,且能对apd-l1可控释放,精准治疗,起到局部治疗全身免疫的效果。
[0087]
本发明氧响应的apd-l1前药可以通过fe3o4纳米颗粒与肿瘤微环境中的h2o2反应产生的
·
oh和原卟啉ppix在660nm激光照射下产生的1o2反应,实现在肿瘤局部的可控释放pd-l1抗体,从而达到特异性免疫激活的目的。
[0088]
尽管本发明的内容已经通过上述优选实施例作了详细介绍,但应当认识到上述的描述不应被认为是对本发明的限制。在本领域技术人员阅读了上述内容后,对于本发明的多种修改和替代都将是显而易见的。因此,本发明的保护范围应由所附的权利要求来限定。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。