1.本发明属于生物医药技术领域,具体而言,涉及一种葡萄糖激酶激动剂及其制备方法和应用。
背景技术:
2.近年来,中国糖尿病呈现出快速增长态势,已从1980年糖尿病发病率0.6%增长到目前的12.8%。国家卫健委最新公布数据表明,我国糖尿病人群已达1.14亿人,其中2型糖尿病占糖尿病人群的近90%。糖尿病并发症涉及血管、眼、肾、足等多个器官,因严重并发症所致的致残、致死率高,严重影响患者健康,给个人、家庭和社会带来沉重的负担。糖尿病是一种慢性代谢性疾病,尽管目前糖尿病治疗方式繁多,在患者长期的血糖控制中,常常会联用2~3种药物使血糖得到充分控制,尽管如此,糖尿病人血糖控制的达标率仍然不高,需要针对新靶点的药物或研发新的治疗方法。
3.葡萄糖激酶(glucokinase, gk)是己糖激酶同工酶中的一种,也是糖代谢过程中一个关键的限速酶,主要分布在胰岛和肝细胞内,gk可以把进入细胞的葡萄糖转变为6磷酸葡萄糖,是肌体葡萄糖的感受器,进餐后血糖升高,超过gk的阈值时,gk就会被激活,启动体内降糖连锁反应,包括在胰岛可促进胰岛素分泌,在肝脏可改善肝糖代谢。gk失去功能突变是单基因糖尿病的致病原因之一,又被称为青年发病的成年型糖尿病2型(mody2)。gk活性降低也是2型糖尿病的致病原因之一,有研究发现通过外源性的药物激活gk后能达到促进胰岛素分泌和增强肝脏清除血中葡萄糖的能力,从而发挥降糖作用,因此活化gk被作为潜在的2型糖尿病的治疗策略。gk激动剂在胰岛可以改善葡萄糖刺激的胰岛素分泌,在肝脏,gk激动剂促进肝糖原合成,从而降低血糖,维持血糖稳态。
4.基于以上,期待一种有效的gka在治疗糖尿病药物中的新应用,为缓解糖尿病的发生和发展带来新的治疗手段。
技术实现要素:
5.针对上述问题,本发明提供了一种葡萄糖激酶激动剂及其制备方法和应用。本发明的葡萄糖激酶激动剂制备方法简单,产率高,得到的产物能够有效降低血糖。
6.本发明的目的及解决其技术问题是采用以下技术方案来实现的。
7.本发明的一个方面提供了一种葡萄糖激酶激动剂,具有如式i的结构式:
i其中,r1选自苯并噻唑基、5-甲基吡啶基、n-甲基吡唑基、6-氟苯并噻唑基中的任一种;r2为氢,其中,所述r1的结构式分别为、、、。
8.本发明的目的及解决其技术问题还通过采用以下技术方案来实现。
9.本发明的另一个方面提供了一种制备葡萄糖激酶激动剂的方法,该方法包括以下步骤:s1:将亮氨酸衍生物与有机物溶解在二氯甲烷中,然后加入1-羟基苯并三唑、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐和n,n-二异丙基乙胺混合并搅拌,然后加入二氯甲烷稀释反应液,加水萃取得第一有机相和第一水相,将第一水相加乙酸乙酯萃取,得第二有机相和第二水相,将第一有机相和第二有机相经干燥、过滤、浓缩后进行柱层析纯化得到第一产物;s2:0℃下,将上述第一产物溶解于二氯甲烷中,然后加入三氟乙酸搅拌反应,结束后在溶液中加入乙酸乙酯,先用水萃取然后调节水相ph>8再用乙酸乙酯萃取,将乙酸乙酯相经干燥、过滤、浓缩得到第二产物;s3:将上述第二产物和邻苯二甲酸酐溶解在乙酸中并搅拌,减压蒸馏去除反应液,用乙酸乙酯和饱和碳酸氢钠溶液萃取,有机相经过滤、蒸发、浓缩后进行柱层析纯化,得到终产物。
10.优选地,步骤s1中所述亮氨酸衍生物为boc-d-亮氨酸或者n-叔丁氧羰基-亮氨酸一水合物。
11.优选地,步骤s1中所述有机物选自2-氨基苯并噻唑、2-氨基-5-甲基吡啶、n-甲基-3-氨基吡唑、2-氨基-6-氟苯并噻唑中的任一种。
12.优选地,步骤s1中所述亮氨酸衍生物与所述有机物的摩尔比为1:1,所述1-羟基苯并三唑、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、n,n-二异丙基乙胺的摩尔比为1:3:6。
13.优选地,步骤s1中所述搅拌条件为:温度为室温,时间为2-6h。
14.优选地,步骤s2中所述搅拌条件为:温度为室温,时间为2-24h。
15.优选地,步骤s3中所述第二产物和邻苯二甲酸酐的摩尔比为1:1。
16.优选地,步骤s3中所述搅拌条件为:温度为120℃,时间为4h。
17.本发明的目的及解决其技术问题还通过采用以下技术方案来实现。
18.本发明的另一个方面提供了葡萄糖激酶激动剂的应用,所述的葡萄糖激酶激动剂为上述的葡萄糖激酶激动剂或者根据上述的制备方法得到的葡萄糖激酶激动剂,所述的应用包括在制备糖尿病治疗药物中的应用,及葡萄糖激酶受损相关糖代谢异常疾病中的应用,或通过激活葡萄糖激酶改善葡萄糖代谢相关疾病领域的应用。
19.借由上述技术方案,本发明至少具有下列优点:1、本发明采用的方法是通过虚拟高通量药物筛选技术,用分子模拟手段计算化合物库中的分子与靶标gk变构调节位点的结合能力,筛选出可能有效的候选化合物,化学合成出来后,再对候选化合物进行gk酶动力学检测,验证其对gk活性的影响。通过对候选化合物反复修饰合成、gk酶动力学检测,找出对gk活性有刺激作用的化合物后,再通过胰岛灌流实验确认该化合物对动态胰岛素分泌的影响,进而在野生型及高脂高糖饮食诱导的肥胖及糖尿病小鼠活体水平确认这些可以激活gk的化合物有降低血糖的作用。
20.2、本发明的制备方法易于操作,所得到的化合物产率高。
21.3、本发明通过化学合成法得到了五种不同结构的化合物,其均以b亮氨酸衍生物为原料,通过与不同的有机物反应得到五种结构不同的化合物,通过实验可知,本发明的五种化合物能够通过提高gk酶活性,促进葡萄糖刺激的胰岛素分泌,调节肝糖原代谢,从而降低血糖。
22.4、本发明的化合物在改善葡萄糖刺激的胰岛素分泌中的特点为,在一定浓度范围,以增加胰岛素分泌的最大值同时不改变葡萄糖的刺激阈值为主要特点,表现为对葡萄糖刺激的胰岛素分泌的促进作用是葡萄糖浓度依赖的,葡萄糖浓度低时,本发明的化合物对胰岛素分泌没有促进作用,避免应用中可能造成的低血糖的问题。
23.上述说明仅是本发明技术方案的概述,为了能够更清楚了解本发明的技术手段,并可依照说明书的内容予以实施,以下以本发明的较佳实施例详细说明如后。
附图说明
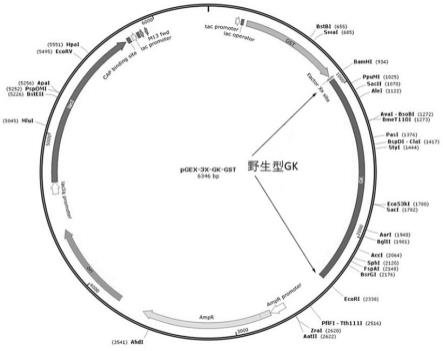
24.图1为野生型gk表达质粒;图2为r447w突变gk表达质粒;图3为根据本发明实施例1得到的产物ar-gk-01对人源gk酶活性的影响曲线图;图4为根据本发明实施例2得到的产物ar-gk-05对人源gk酶活性的影响曲线图;图5为根据本发明实施例3得到的产物ar-gk-08对人源gk酶活性的影响曲线图;图6为根据本发明实施例4得到的产物ar-gk-10对人源gk酶活性的影响曲线图;图7为根据本发明实施例5得到的产物ar-gk-25对人源gk酶活性的影响曲线图;图8为通过分别添加1μm、2μm、10μm的ar-gk-01的情况下,葡萄糖浓度对gk酶活性影响曲线图;图9为通过分别添加0.5μm、1μm、2μm、5μm的ar-gk-05的情况下,葡萄糖浓度对gk酶活性影响曲线图;
图10为通过添加5
ꢀµ
m ar-gk-08的情况下,葡萄糖浓度对gk酶活性影响曲线图;图11为与野生型gk相比,葡萄糖浓度对单基因糖尿病的gk突变(r447w)酶活性影响曲线图;图12为通过添加10
ꢀµ
m ar-gk-01的情况下,葡萄糖浓度对野生型和单基因糖尿病的gk突变(r447w)酶活性影响曲线图;图13为与空白对照相比,通过添加10
ꢀµ
m ar-gk-01的情况下,葡萄糖浓度单基因糖尿病的gk突变(r447w)酶活性影响曲线图;图14为0.1
ꢀµ
m和0.5
ꢀµ
m的ar-gk-01对葡萄糖刺激胰岛素分泌(gsis)的影响曲线图;图15为2
ꢀµ
m和10
ꢀµ
m的ar-gk-01对葡萄糖刺激胰岛素分泌(gsis)的影响曲线图;图16为0.5
ꢀµ
m的ar-gk-01和对标葡萄糖激酶激动剂mk-0941对葡萄糖刺激胰岛素分泌(gsis)影响的比较;图17为ar-gk-01和对标葡萄糖激酶激动剂mk-0941的剂量爬坡对2.8 mm葡萄糖刺激胰岛素分泌(gsis)影响的比较;图18为ar-gk-01和对标葡萄糖激酶激动剂mk-0941的剂量爬坡对3.5 mm葡萄糖刺激胰岛素分泌(gsis)影响的比较;图19为ar-gk-01对野生型小鼠的血糖随时间改变的影响曲线图;图20为ar-gk-01对高脂饮食诱导的肥胖或糖尿病小鼠的血糖随时间改变的影响曲线图;图21为糖原标准曲线;图22为ar-gk-01在剂量为20 mg/公斤体重时,对野生型小鼠及sur1敲除小鼠肝糖原的影响;图23为灌胃ar-gk-01(20mg/公斤体重)后,小鼠血浆ar-gk-01浓度变化曲线图。
具体实施方式
25.为了使本发明实现的技术手段、创作特征、达成目的与功效易于明白了解,下面将结合本发明实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有作出创造性劳动前提下所获得的所有其它实施例,都属于本发明保护的范围。
26.根据本发明所述的葡萄糖激酶激动剂,其具有如式i的结构式:
。其中,r1选自苯并噻唑基、5-甲基吡啶基、n-甲基吡唑基、6-氟苯并噻唑基中的任一种;r2为氢;其中,所述r1的结构式分别为、、、。
27.根据本发明所述的制备葡萄糖激酶激动剂的方法,包括以下步骤:s1:将亮氨酸衍生物与有机物溶解在二氯甲烷中,然后加入1-羟基苯并三唑、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐和n,n-二异丙基乙胺混合并搅拌,然后加入二氯甲烷稀释反应液,加水萃取得第一有机相和第一水相,将第一水相加乙酸乙酯萃取,得第二有机相和第二水相,将第一有机相和第二有机相经干燥、过滤、浓缩后进行柱层析纯化得到第一产物;亮氨酸衍生物为boc-d-亮氨酸或者n-叔丁氧羰基-亮氨酸一水合物;有机物选自2-氨基苯并噻唑、2-氨基-5-甲基吡啶、n-甲基-3-氨基吡唑、2-氨基-6-氟苯并噻唑中的任一种;亮氨酸衍生物与所述有机物的摩尔比为1:1,所述1-羟基苯并三唑、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、n,n-二异丙基乙胺的摩尔比为1:3:6;搅拌条件为:温度为室温,时间为2-6h;s2:0℃下,将上述第一产物溶解于二氯甲烷中,然后加入三氟乙酸搅拌反应,结束后在溶液中加入乙酸乙酯,先用水萃取然后调节水相ph>8再用乙酸乙酯萃取,将乙酸乙酯相经干燥、过滤、浓缩得到第二产物;搅拌条件为:温度为室温,时间为2-24h;s3:将上述第二产物和邻苯二甲酸酐溶解在乙酸中并搅拌,减压蒸馏去除反应液,用乙酸乙酯和饱和碳酸氢钠溶液萃取,有机相经过滤、蒸发、浓缩后进行柱层析纯化,得到终产物;第二产物和邻苯二甲酸酐的摩尔比为1:1;搅拌条件为:温度为120℃,时间为4h。
28.根据上述描述得到的通用合成路线为:
167.51 (2c), 159.87 (1c), 158.20 (1c), 157.49 (1c), 134.63 (2c), 131.71 (2c), 123.29 (2c), 114.40 (1c), 114.16 (1c), 108.33 (1c), 108.06 (1c), 50.96 (1c), 36.95 (1c), 24.44 (1c), 23.21 (1c), 21.02 (1c)。
46.实施例6 gk酶动力学检测1、gk蛋白的表达与纯化1.1、gk蛋白的表达(1)原核表达体系的构建:目的基因按照标准程序合成,使用大肠杆菌表达载体质粒pgex-3x:a.在酶切位点bamhⅰ/ecorⅰ处插入野生型gk蛋白基因构建重组表达载体pgex-3x-gk-gst,氨苄青霉素(amp)抗性基因筛选,带有gst标签蛋白。质粒图及gk基因插入位置见图1。
47.b.gk突变型(r447w)质粒构建:野生型gk原核表达质粒pgex-3x-gk-gst按照点突变标准程序进行点突变,构建gk突变型r447w质粒pgex-3x-r447w-gst, 酶切位点bamhⅰ/ecorⅰ, 氨苄青霉素(amp)抗性基因,带有gst标签蛋白。质粒图及突变gk插入位置见图2。
48.(2)转化到表达菌a.在超净工作台将2微升pgex-3x-gck质粒加入50
ꢀµ
l bl21 (de 3)感受态中混合,冰浴30 min。
49.b.热激:将混合物放入水浴锅,42℃水浴热激90s后迅速拿出。
50.c.冰浴3 min后,在超净工作台向混合物加入900微升无抗生素的液体lb培养基并放到37℃摇床,220 rmp转速摇45 min。
51.d.2000 rmp转速离心2 min。
52.e.在超净工作台弃大部分上清,留大约50微升培养液重悬菌体,吸出加入有氨苄青霉素(amp)抗生素(1 mg/ml)的固体lb培养板上,滚珠涂布法将菌液涂布均匀,倒置放入37℃培养箱中过夜培养。
53.(3)诱导表达a.在超净工作台中操作,从培养板中挑选单菌落到加入5 ml 含有amp抗生素(1 mg/ml)的液体lb培养基,37℃,220 rmp转速的摇床上过夜培养。
54.b.在超净工作台中操作,将过夜培养的菌液转接到含有200ml液体lb培养基的锥形瓶中,加入amp抗生素,37℃,220 rmp转速摇床上培养。
55.c.培养3-4h左右时,从中吸取1 ml菌液检测od600值,直到od600在0.6-0.8之间,加入诱导剂iptg(100 mm)按照1:1000的比例加入锥形瓶中进行诱导表达,37℃,220 rmp转速培养12h左右。
56.d.收集菌液:4℃,12000 rmp,离心20 min,弃上清。pbs洗涤一遍,44℃,12000 rmp,离心20 min,弃上清。
57.1.2、gk蛋白的纯化重组蛋白gk携带gst标签,使用碧云天gst标签蛋白纯化试剂盒纯化蛋白。
58.a.按照每克细菌沉淀湿重加入4 ml (2-5 ml均可) 的比例加入裂解缓冲液,充分重悬菌体。
[0059] b.加入溶菌酶至终浓度为1 mg/ml并混匀,冰水浴或冰上放置30 min。
[0060]
注:溶菌酶可以用裂解液配制成100 mg/ml的母液,临时用前加入。溶菌酶配制成母液后,可以适当分装后-20
º
c保存。
[0061]
c.冰上超声裂解细菌。超声功率200-300w,每次超声处理10 s,每次间隔10 s,共超声处理6次。
[0062]
d.4
º
c 10,000g离心20-30 min,收集细菌裂解液上清并置于冰水浴或冰上,可以取20微升上清留作后续检测用。
[0063]
e.取1 ml混合均匀的beyogold
™ꢀ
gst-tag purification resin,4
º
c离心 (1000 g
×
10 s)弃去储存液,向凝胶中加入0.5 ml裂解缓冲液以重悬并平衡凝胶,4
º
c离心 (1000 g
×
10 s)弃去液体,再重复平衡1~2次,弃去液体。将约4 ml细菌裂解液上清加入其中,4
º
c在侧摆摇床或水平摇床上缓慢摇动60 min。
[0064]
f.将裂解液和beyogold
™ꢀ
gst-tag purification resin的混合物装入本试剂盒提供的亲和层析柱空柱管 (3毫升) 中。注:也可先取1 ml混合均匀的50% beyogold
™ꢀ
gst-tag purification resin装柱,然后用0.5 ml裂解缓冲液平衡2~3次后加入约4 ml细菌裂解液上清,后续可以把流穿液收集后重复上柱3~5次以充分结合目的蛋白。
[0065]
g.将纯化柱底部的盖子打开,在重力作用下使柱内液体流出,收集约20微升流穿液作后续分析用。
[0066]
h.洗柱5次,每次加入0.5~1 ml裂解缓冲液,每次均收集约20微升穿柱的液体用于后续的分析检测用。洗柱及下一步洗脱过程中可以用bradford法简单快速地检测每次洗涤液和洗脱液中的蛋白含量,从而考虑增加或减少洗涤和洗脱的次数。
[0067]
i.洗脱目的蛋白6-10次,每次用0.5 ml洗脱缓冲液。将每次的洗脱液分别收集到不同的离心管中。收集获得的洗脱液即为纯化的gst标签蛋白样品。
[0068]
注:洗脱缓冲液的组分为50 mm tris, 150 mm nacl, 10 mm gsh, ph 8.0。
[0069]
1.3、gk酶动力学实验(1)溶液配置及蛋白浓度测定:a.分别配置以下浓度的工作试剂,即mgcl2: 6 mm; bsa: 0.1%; kcl: 150 mm; hepes: 100 mm; nadp
: 1 mm; g-6-pdh: 5 unit/ml; dtt: 2 mm;atp: 5 mm;b.配置10 ml预混液,即1 ml mgcl2(60 mm) 1 ml bsa(1%) 1.5 ml kcl(1 mm) 1 ml hepes(1 m) 1 ml nadp
(10 mm) 10
ꢀµ
l g-6-p-gdh(5unit/ml) 0.2 ml atp(250 mm) 20
ꢀµ
l dtt(1 m) 4.27 ml h2o,ph 7.4。将预混液在37℃水浴锅中加热5分钟,以进行gk酶稀释。
[0070]
c.按照蛋白浓度测定试剂盒(碧云天,p0007)说明书方法测定出样品的蛋白浓度。
[0071]
d.测定化合物对gk酶活性的作用。
[0072]
e.wt-gst-gk浓度:按照预实验的浓度摸索,用预混液进行稀释。
[0073]
f.待测化合物浓度:将化合物稀释到1 mm,然后再加入96孔板中用预混液进行倍比稀释;g.反应体系(100微升):(2)葡萄糖剂量曲线:10微升待测化合物(固定浓度,根据试验要求调整) 40 微升预混液 40 微升 葡萄糖(不同浓度) 10 微升wt-gst-gk(通过预混液稀释)
(3)化合物有效性检测:40微升葡萄糖(工作浓度5 mm) 40微升预混液 10微升待测化合物 (不同浓度) 10微升wt-gst-gk(通过预混液稀释);(4)检测:低速离心混匀后,37℃恒温箱中孵育30 min,放入酶标仪中检测,在室温下,设置检测波长为340 nm每隔30秒读取,持续25 min。
[0074]
(5)分析数据:将数据导出至excel表格后,用最后一个循环的数值减去初始值,得到不同浓度药物的对应数值输入graphpad prism 8软件中进行分析,使用allosteric sigmoidal或log(agonist) vs. response方程的回归线拟合对酶动力学参数进行估算分析。
[0075]
1.4、实验结果(1)五种化合物对gk的激动效果gk激动剂化合物共5个,包括ar-gk-01、ar-gk-05、ar-gk-08、ar-gk-10、 ar-gk-25,分别针对gk酶动力学进行了检测,方式为固定葡萄糖浓度(5 mm)的情况下,检测不同化合物浓度剂量曲线,结果见图3~7。由图3-7的结果可知,ar-gk-01、ar-gk-05、ar-gk-08、ar-gk-10、ar-gk-25对gk酶有激动作用。
[0076]
因各个化合物对gk的激动效果不同,选择了ar-gk-01、ar-gk-05和ar-gk-08这3个化合物的单一或不同浓度进行葡萄糖剂量曲线检测,即固定化合物浓度,检测不同浓度葡萄糖下的酶动力改变,进一步确定化合物效果,结果见图8~10。结果发现化合物ar-gk-01浓度在1
ꢀµ
m、2
ꢀµ
m、10
ꢀµ
m时酶促反应达最大速度一半时的底物(葡萄糖)浓度s
0.5
分别为3.155 mm、2.633 mm和1.355 mm,化合物ar-gk-05浓度在0.5
ꢀµ
m、1
ꢀµ
m、2
ꢀµ
m和5
ꢀµ
m时酶促反应达最大速度一半时的底物(葡萄糖)浓度s
0.5
分别为7.733 mm、6.698 mm、5.534 mm、3.364 mm,化合物ar-gk-08浓度在5
ꢀµ
m时酶促反应达最大速度一半时的底物(葡萄糖)浓度s
0.5
分别为3.163 mm。由此说明ar-gk-01、ar-gk-05和ar-gk-08对gk活性均有显著的刺激作用,且呈浓度梯度依赖效应,其中ar-gk-01的刺激作用更强,为此次试验中筛选出的最优化合物。
[0077]
(2)ar-gk-01对mody2致病基因突变(r447w)gk的激动作用:为了验证gk激动剂ar-gk-01是否对引起单基因糖尿病的突变gk也有激动作用,我们选择了r447w突变gk进行表达及蛋白纯化,并研究了突变gk的酶动力学改变。
[0078]
如图11所示,与正常野生型gk比较,r447w突变gk的葡萄糖s
0.5
明显升高,从同时检测的配对试验正常gk的8.99 mm升高到17.04 mm,提示r447w突变致gk酶活性受损。
[0079]
如图12所示,10
ꢀµ
m的ar-gk-01纠正了受损的gk活性,使得r447w突变葡萄糖的s
0.5
从17.04 mm葡萄糖降低到5.317 mm,野生型gk则在10
ꢀµ
m的ar-gk-01的刺激下,葡萄糖的s
0.5
从8.99 mm葡萄糖降低到3.898 mm,说明ar-gk-01不仅可以刺激正常野生型gk活性,同样对酶功能受损的突变gk也有激动作用。
[0080]
如图13所示,10
ꢀµ
m的ar-gk-01不仅降低了r447w突变gk的葡萄糖的s
0.5
,同时也提升了r447w突变gk的vmax,改善了h指数,说明ar-gk-01可以全方位修复因突变致功能受损的gk,可以用于治疗gk失去功能突变说致的单基因糖尿病。
[0081]
实施例7 gk激动剂对葡萄糖刺激胰岛素分泌的影响研究(1)胰岛分离a.小鼠麻醉后固定于手术板上,打开腹腔,用止血钳闭锁胆管进入十二指肠的入
口,之后用5 ml注射器及31.5号针头将2 mg/ml的胶原酶溶液注射进入胆管,至胰腺充分充起后停止。
[0082]
b.剥离胰腺,清理掉脂肪及非胰腺组织后,将胰腺转移到50 ml离心管内,再倒入3 ml的2 mg/ml胶原酶溶液,在37℃水浴中震荡4~5分钟。
[0083]
c.震荡结束后,立刻在50 ml离心管内加入hanks缓冲液至50 ml,离心管转速为2000 rpm,离心完毕后移走上清,剩余组织沉淀加入5 ml histopaque-1119溶液,震荡混匀。
[0084]
d.缓慢加入5 ml histopaque-1077溶液,再缓慢加入5 ml hanks缓冲液,离心管转速为2000 rpm。
[0085]
e.从分层的液体间移走胰岛组织,在解剖显微镜下挑取纯化胰岛,清洗后培养。
[0086]
(2)胰岛培养。
[0087]
a.rpmi1640培养液的配制:葡萄糖10 mm,谷氨酰胺2 mm,胎牛血清10%,碳酸氢钠2 g/l,青霉素100 units/ml,链霉素10
ꢀµ
g/ml,调节ph到7.2。
[0088]
b.用rpmi1640培养液将分离纯化的胰岛在37℃、5% co2及95%空气湿度的培养箱内培养1~2天。
[0089]
(3)胰岛灌流实验a.缓冲液配制:115 mm的nacl、24 mm的nahco3、5 mm的kcl、1 mm的mgcl2和2.5 mm的cacl2,调节ph到7.4,再加入0.25%牛血清白蛋白。
[0090]
b.配置25 mm葡萄糖、2.8 mm葡萄糖、3.5 mm葡萄糖的刺激液。c.手工捡取120个大小相同的胰岛,置于胰岛小室内,根据实验需求,置于水浴锅内。
[0091]
d.将配制好的反应液分别置于水浴锅内(37℃),插入对应的进样管,如下表1为灌流程序:表1 灌流程序注释:a为缓冲液,b为刺激液,c为30 mm氯化钾,曲线 1到6为爬坡灌流,a与b逐渐混合,a从100%逐渐降低到0,同时b从0逐渐升高到100%。
[0092]
e.由collector
ꢁ
fraction
ꢁ
waters收集分泌的胰岛素于96深孔板内,设定1 ml/min收集反应液,保存于-20℃冰箱直到激素测定。
[0093]
(4)胰岛素分泌值检测。
[0094]
a.取胰岛灌流实验收集于96深孔板的溶液10 μl/孔,移入384孔板;
b.按照htrf胰岛素测定试剂盒说明加入抗体,震荡混匀后置于室温孵育2小时后,用bmg的clariostar酶标仪htrf程序读数,根据标准曲线计算激素分泌值,通过测定胰岛素分泌来确认刺激物或药物对胰岛素分泌的影响。
[0095]
(5)实验结果。
[0096]
葡萄糖浓度爬坡(浓度增加的速度为0.5 mm/min)刺激的胰岛灌流试验证明,葡萄糖刺激胰岛素分泌(gsis)的曲线特点为,葡萄糖浓度阈值大约在7 mm,vmax保持在10 ng/120个胰岛/min的水平。如图14所示,在gsis试验的同时分别加入浓度为0.1
ꢀµ
m和0.5
ꢀµ
m ar-gk-01后,0.1
ꢀµ
m与空白对照一致,没有促进gsis的作用。ar-gk-01的浓度提升到0.5
ꢀµ
m后,gsis的vmax增加到15 ng/120个胰岛/min的水平,提升大约50%,而阈值没有改变。当ar-gk-01的浓度进一步提升到2
ꢀµ
m后(图15),在不改变gsis阈值的情况下,vmax增加到19 ng/120个胰岛/min的水平,从0.5
ꢀµ
m的ar-gk-01又提升大约40%。但当ar-gk-01浓度升高到10
ꢀµ
m后,gsis的阈值从7 mm降低到4 mm,vmax没有进一步的升高,回到0.5
ꢀµ
m的ar-gk-01的水平(图15),因此,在ar-gk-01为0.1
ꢀµ
m ~2
ꢀµ
m 浓度范围内,胰岛素分泌的最大值随ar-gk-01的浓度增加而增加,但gsis的阈值没有改变,说明ar-gk-01促进gsis在该浓度范围内是葡萄糖依赖的。图16显示ar-gk-01与另一个葡萄糖激酶激动剂mk-0941之间的显著差异,同样的0.5
ꢀµ
m,ar-gk-01主要特点是增加vmax而不改变gsis的葡萄糖阈值,而mk-0941则是显著降低gsis葡萄糖的阈值,同时也降低vmax。
[0097]
为了进一步验证ar-gk-01促进gsis是葡萄糖浓度依赖的特点,选择了两个低葡萄糖浓度下,ar-gk-01对胰岛素分泌的促进作用的试验,同样也对标mk-0941。图17和图18在灌流过程中葡萄糖溶液浓度维持在2.8 mm或3.5 mm时,这个葡萄糖浓度不能刺激胰岛素分泌,胰岛素分泌曲线的基线保持在低水平,在此基础上,行化合物浓度爬坡试验,浓度增加的速度是0.125
ꢀµ
m/min。图17显示在2.8 mm背景葡萄糖的情况下,ar-gk-01即使到最大的 5
ꢀµ
m的水平,也不具备刺激低浓度葡萄糖(2.8 mm)刺激胰岛素分泌的作用,而mk-0941则显著增加胰岛素分泌,刺激阈值为0.5
ꢀµ
m,1.75
ꢀµ
m时刺激作用达到最高。图18显示葡萄糖背景浓度为3.5 mm时的情况,同样,ar-gk-01即使到最大的 5
ꢀµ
m的水平,也不具备明显刺激低浓度葡萄糖(3.5 mm)刺激胰岛素分泌的作用,只有在0.875~1.25
ꢀµ
m之间有轻微的胰岛素分泌增多,但与胰岛素分泌基线比较没有统计学的差异。而mk-0941则显著增加胰岛素分泌,阈值和最大刺激浓度与背景葡萄糖浓度为2.8 mm时一致。说明发挥血糖感受器作用的gk在葡萄糖处于较低水平时不刺激胰岛素分泌,同样ar-gk-01对胰岛素分泌也没有进一步的促进作用,再次说明ar-gk-01发挥作用是葡萄糖依赖的,理论上避免了药物本身引起低血糖的发生的可能性。
[0098]
实施例8 活体动物试验1、正常饮食小鼠口服葡萄糖耐量试验(ogtt)1.1、溶剂配置:(1)ar-gk-01溶液的配置:取6 mg ar-gk-01溶于30 μl dmso,再溶于60 μl蓖麻油,最后用0.5%羧甲基纤维素钠(cmc)定容至3 ml,最终浓度为2 mg/ml。
[0099]
(2)cmc-dmso-蓖麻油混合溶液(溶剂对照):30 μl dmso加入60 μl蓖麻油,然后用0.5%羧甲基纤维素钠(cmc)定容至3 ml。
[0100]
1.2、实验过程:雄性正常(wt)小鼠平均分成两组,上午8:30开始禁食,4小时后,于下午12:30测血糖,之后,实验组灌胃ar-gk-01(剂量为20 mg/kg体重),对照组灌胃cmc-dmso-蓖麻油混合溶液(同容量的溶剂对照),1 小时后测血糖,接着行ogtt,灌胃葡萄糖(剂量2.0 g/kg体重),之后每30 min测一次血糖。
[0101]
2、高脂高糖诱导的肥胖/糖尿病模型及ogtt试验:2.1、高糖高脂诱导的肥胖/糖尿病小鼠模型的制备:通过喂食60%高脂饲料和7.5%蔗糖水,经过4个月喂食后,小鼠体重从26.3 g增加至49.1 g,同时血糖升高,制备成功肥胖/糖尿病小鼠模型。下表2、3分别为正常饲料与高脂饲料的营养成分。
[0102]
表2 正常饲料表3高脂饲料2.2、溶液配置:(1)ar-gk-01溶液的配置:取8 mg ar-gk-01溶于40 μl dmso,再溶于80 μl蓖麻油,最后用0.5%羧甲基纤维素钠(cmc)定容至4 ml。
[0103]
(2)cmc-dmso-蓖麻油混合溶液(溶剂对照):40 μl dmso加入80 μl蓖麻油,然后用0.5%羧甲基纤维素钠(cmc)定容至4 ml。
[0104]
2.3、实验过程:雄性肥胖小鼠分成两组,上午9:00开始禁食,4小时后,于下午13:00测血糖,实验组灌胃ar-gk-01(20 mg/kg体重),对照组灌胃cmc-dmso-蓖麻油混合溶液(同容量溶剂对照),1 小时后测血糖,接着行ogtt,灌胃葡萄糖(剂量2.0 g/kg体重),之后每30 min测一次血糖。
[0105]
3、试验结果:图19显示ar-gk-01在剂量为20 mg/kg体重的条件下对正常小鼠ogtt的改善作用,灌胃葡萄糖后的30 min,60 min,90 min和150 min的血糖用药组明显低于溶剂对照组,提
示ar-gk-01可以改善葡萄糖耐量。图20显示ar-gk-01对高脂高糖饮食诱导的肥胖/糖尿病小鼠葡萄糖耐量的改善作用,首先肥胖/糖尿病小鼠空腹4小时的血糖值明显升高,肥胖/糖尿病小鼠的血糖为10.9
±
0.5 mm (n=15),正常小鼠空腹4小时血糖为8.8
±
0.4 mm (n=11),p《0.01。ar-gk-01在剂量为20 mg/kg体重的条件下对肥胖/糖尿病小鼠的ogtt有明显的改善作用,表现为ogtt的60 min,90 min,120 min,150 min,180 min和210 min的血糖都明显低于溶剂对照组,说明ar-gk-01对肥胖/糖尿病小鼠葡萄糖耐量有明显的改善。
[0106]
实施例9 ar-gk-01对肝糖原代谢的影响1、小鼠肝糖原检测方法:1.1、试剂和仪器1)amyloglucosidase(sigma): 15 u/ml2)glucose oxidase ( macklin ): 200 u/ml3)hrp (solarbio): 100 u/ml4)abts ( macklin ): 1 mm5)50 mm sodium phosphate (ph 7.4/ph 4.5)6)naoh: 0.2 n7)tca: 5%、10%8)96孔板9)clariostar 多功能酶标仪10)预混液的配置:10 μl 100 u/ml hrp储备溶液和 20 μl 200 u/ml glucose oxidase储备溶液至 970 μl 50 mm sodium phosphate(ph 7.5)配制成1 ml 。
[0107]
2、肝脏糖原的提取和检测2.1、提取糖原1)称取冻存或新鲜小鼠肝脏约50 mg置于1.5ml尖底ep管中,加入50μl 10% tca和200 μl 5% tca, 使用组织匀浆器在冰上进行匀浆,彻底匀浆后使用离心机进行离心,5000 rpm,10 min。
[0108]
2)离心后取上清置于ep管中,沉淀暂存于4℃。上清加入等体积预冷的95%乙醇溶液,充分混匀后静置10 min(也可延长时间或4℃过夜),使用离心机进行离心,5000 rpm,10 min。
[0109]
3)离心后弃上清,吸取残留上清并干燥沉淀,加入200 μl超纯水溶解沉淀,沸水浴2 min。
[0110]
2.2. 糖原标准曲线:1)通过在50 mm sodium phosphate(ph 4.5)稀释 4 mg/ml糖原标准品(在使用前制备新鲜的糖原标准品),倍比稀释,每个浓度取50 μl置于ep管中,在每个管中加入5 μl的 15 u/ml amyloglucosidase (sigma),将样品在55℃恒温下反应20 min。
[0111]
2)将步骤1中的反应物分别取30 μl置于96孔板中,加入同体积的预混液,混合均匀后再37℃恒温箱中孵育45 min。
[0112]
3)取20 μl孵育液加入100 μl 1 mm abts,37℃恒温箱30 min。
[0113]
4)在clariostar 多功能酶标仪中,检测波长为405 nm处吸光度的值来测定糖原含量,graphpad prism 软件中进行分析,使用方程的回归线拟合进行分析得到反应曲线如
图21。2.3、肝糖原检测:1)将提取的糖原样品使用50mmsodiumphosphate(ph4.5)溶液进行稀释一定倍数后取50μl置于ep管中,在每个管中加入5μl的15u/mlamyloglucosidase(sigma),将样品在55℃恒温下反应20min。
[0114]
2)将步骤1中的反应物分别取30μl置于96孔板中,加入同体积的预混液,混合均匀后再37℃恒温箱中孵育45min。
[0115]
3)取20μl孵育液加入100μl1mmabts,37℃恒温箱30min后使用酶标仪(405nm)读取结果。
[0116]
4)将每个样品的δod与标准曲线进行比较,以确定和推断样品中存在的糖原(仅使用标准曲线范围内的值)。
[0117]
2.4、肝脏糖原含量计算方式肝脏糖原含量与肝脏湿重将样品经过糖原标曲得出的结果与肝脏湿重进行计算:总肝糖原(mg/g湿重)=糖原(mg)/组织重量(g)3、动物试验:3.1、2mg/mlar-gk-01溶液配制:取20mgar-gk-01溶于100μldmso,再溶于200μl蓖麻油,最后用0.5%羧甲基纤维素钠(cmc)定容至10ml。
[0118]
3.2、实验过程:准备5只wt小鼠和5只sur1-敲除小鼠,每天早10:00测血糖后灌胃ar-gk-01(20mg/kg),连续3天。第三天下午13:00处死取肝脏组织,提取肝糖原并检测含量。另各取5只正常饲养的wt和sur1敲除小鼠,于下午13:00处死取肝脏组织作为对照,提取肝糖原并检测含量。
[0119]
3.3、试验结果:如图22所示,ar-gk-01在剂量为20mg/公斤体重时,对野生型小鼠(溶剂对照组n=6,给药组n=4)及sur1敲除小鼠(sur1-ko,溶剂对照组n=6,给药组n=4)肝糖原的影响。ar-gk-01用药3天对wt小鼠的肝糖原没有影响。sur1-ko小鼠的肝糖原明显高于wt小鼠,ar-gk-01降低了sur1-ko小鼠的肝糖原,使其恢复到wt小鼠的水平,说明ar-gk-01对肝糖原代谢有影响。
[0120]
实施例10ar-gk-01小鼠体内药物代谢和组织分布检测1、2mg/mlar-gk-01溶液配制:取20mgar-gk-01溶于100μldmso,再溶于200μl蓖麻油,最后用0.5%羧甲基纤维素钠(cmc)定容至10ml。
[0121]
2、实验过程:药物代谢实验:3只c57bl/6j雄性8周龄小鼠过夜饥饿后,口服给予2mg/ml的ar-gk-01溶液,给药体积为100μl/10g小鼠体重,分别于给药前、给药后15、30、60、120、240以及480分钟后,眼眶取血100μl,放置于抗凝管中,12000rpm,离心2分钟,取出上层血浆,保存于-80度冰箱用于检测药物浓度。
[0122]
组织分布:3只c57bl/6j雄性8周龄小鼠过夜饥饿后,口服给予2 mg/ml的ar-gk-01溶液,给药体积为100 μl/10 g 小鼠体重,给药后60分钟后,将胰腺以及肝脏组织取出,在生理盐水中将残留血液清洗干净后,放置于冻存管中,冻存于-80度冰箱用于检测药物浓度。
[0123]
3、ar-gk-01的lc/ms/ms分析方法3.1、样品预处理方法1)、血浆样品:向20
ꢀµ
l血浆中加入200
ꢀµ
l meoh/acn (1/1,v/v),涡流混匀后离心(15000rpm,5min),取20
ꢀµ
l上清液 20
µ
l acn/h2o (1/1,v/v) 混匀后进样分析。
[0124]
2)、组织样品:组织称重后,按重量加入5倍体积meoh/can (1/1,v/v)匀浆,涡流混匀后离心(15000rpm, 5min),获得组织匀浆液,取40
ꢀµ
l上清液进样分析。
[0125]
3.2、色谱方法色谱柱:waters acquity uplcr beh phenyl 1.7
ꢀµ
m(2.1*50 mm,1.7
ꢀµ
m)流速:0.5 ml/mina:5mmnh4oacinh2owith0.1�b:0.1�inacn/meoh(9/1,v/v)梯度如下表4所示表4梯度自动进样器温度:10
°
c柱温:45
°
c3.3、质谱方法质谱参数如表5所示。
[0126]
表5 质谱参数
4、实验结果:如表6所示,三只小鼠用药后不同时间的ar-gk-01的血药浓度(ng/ml),0.25小时即可见血药浓度显著升高,峰值出现在0.5小时,之后浓度逐步降低,只8小时仍然有药物残留,见图23。表7显示药代动力学参数,ar-gk-01的半衰期是在2.93小时,达峰时间是0.33小时,在剂量为20 mg/kg体重的情况下,最大血药浓度为1273 ng/ml。表8显示肝脏和胰腺的组织分布,在肝和胰腺均有分布。
[0127]
表6.不同时间中小鼠血浆药物浓度(ng/ml)
以上所述,仅是本发明的较佳实施例而已,并非对本发明作任何形式上的限制,虽然本发明已以较佳实施例揭露如上,然而并非用以限定本发明,任何熟悉本专业的技术人员,在不脱离本发明技术方案范围内,当可利用上述揭示的方法及技术内容作出些许的更动或修饰为等同变化的等效实施例,但凡是未脱离本发明技术方案的内容,依据本发明的技术实质对以上实施例所作的任何简单修改、等同变化与修饰,均仍属于本发明技术方案的范围内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。