测定mrna平均分子量以及cap0/1、修饰核苷酸及氧化物的方法
技术领域
1.本发明属于生物医药领域,具体涉及一种测定样品中mrna平均分子量的方法及其应用,所述应用特别涉及一种测定mrna平均分子量以及cap0/1、修饰核苷酸及氧化物的方法。
背景技术:
2.随着核酸药物的发展,mrna被视为了一种可以用于药物制造的新选择。1990年,一段mrna被注射进入小鼠体内,并成功编码出了蛋白质。这段mrna则是通过一种名为体外转录的技术得到的。随后,一项1992年的研究发现注射抗利尿激素编码mrna可以成功诱导大鼠的下丘脑的神经活动。虽然mrna显示出很好的生物活性,但是受限于其自身的不稳定性,强免疫原性和体内递送困难,mrna远远无法应用于临床疾病治疗中。
3.随着过去的十几年中技术的不断发展和研究的不断深入,mrna逐渐成为了一种拥有广阔应用前景的治疗手段,特别是在疫苗和蛋白替代疗法方面优势尤为突出。在抗击病毒感染性疾病方面,mrna显示出了如下一些优势:
4.1.mrna是一个没有传染性也没有整合能力的平台,不会有任何潜在的感染风险,也不会发生突变性的整合。并且mrna会随着细胞代谢逐步降解,它自身的半衰期可以多种修饰和递送手段来进行控制。mrna自身固有的免疫原性也可以进行调节以提高用药的安全性。
5.2.多种修饰方法可以让mrna变得更加稳定也更容易递送。高效的mrna递送可以通过载体分子的装载迅速进入细胞质内实现高效表达。mrna是一种极小型的遗产信息载体。所以必须避免免疫系统的清除。而mrna可以随心所欲的进行快速设计。
6.3.mrna可以通过高效率的体外转录技术,实现低成本高效率的制造。
7.现在mrna疫苗领域发展速度极快,在过去的几年中产生了非常多的临床前研究数据。
8.也有不少项目开展了临床实验。所有的这一切都表明mrna疫苗非常有潜力迎接来自于各种感染性疾病和癌症的挑战。
9.在dna翻译和和核糖体蛋白组装的进程中,mrna恰好充当了中间媒介。现在用于疫苗开发的mrna主要分为两大类:由病毒衍生出的非复制型mrna和自扩增rna。一般的非复制型mrna疫苗包含目的抗原编码序列,5
′
和3
′
utr。而自扩增rna不光拥有抗原编码序列,还有可以实现细胞内自我复制提高蛋白表达水平的病毒复制调节序列。
10.临床上使用的mrna是通过体外转录(ivt)技术获得的。简单来说体外转录rna是利用线性化的dna模板利用t7,t3或者sp6等rna聚合酶合成得到的。得到的转录产物包含蛋白编码序列,两侧的非编码序列utr,5
′
端帽子和poly a尾巴。这种体外合成的mrna完全类似于真核细胞细胞质中成熟的mrna分子。
11.mrna体内递送的复杂性也越来越被人们所认识。裸露的mrna会被细胞外的mrna酶
迅速降解掉,无法进入细胞内发挥功能。所以大量的递送载体试剂被开发用于mrna的细胞内吞并保护mrna不被降解。一旦mrna进入细胞质,细胞就会启动翻译机制并进行有序折叠制造有功能的蛋白。mrna的这些特性非常有利于疫苗和蛋白替代疗法的开发。因为这两种药物需要将正确的蛋白质递送至细胞内部发挥功能。所有这些体外转录合成的mrna都会在正常的生理过程中被降解,这样也大大降低了代谢毒性。
12.近些年来很多的公司及研究机构进行了mrna的研究开发,尝试降低mrna自身的免疫原性提要成药行。通过对mrna的序列进行优化可以使其更容易导入。使用高效无毒的递送载体也可以使mrna在细胞内高效表达目的抗原。为了更有效的启动免疫应答机制,很多新型的辅剂也被研发出来用于提高mrna疫苗的有效性。
13.mrna的翻译效率和稳定性是十分重要的。位于蛋白编码序列两侧的5
′
和3
′
utr直接影响mrna的翻译效率和稳定性,从而直接决定了疫苗的效果。这部分可以通过引入从病毒到真核生物的不同序列来极大的提高药用mrna的半衰期和表达效率。5
′
cap结构也是mrna高效表达所必须的部分。在体外转录的过程中通过牛痘病毒加帽酶可以为mrna加上cap结构。当然这种结构也可以使用有机合成品或者反向cap类似物来进行添加。poly a尾巴在调节mrna功能方面也起着重要作用,所以一个被优化过的poly a需要通过模版或者加尾酶添加进mrna。不同密码子的运用也可以影响蛋白的表达。将稀有密码子改变为常用的同义密码子trna也是一种提高蛋白翻译效率的常用方法。也有研究表明提高gc碱基丰度同样也可以提高mrna的体外稳定性和体内蛋白翻译效率。
14.尽管通过改变mrna的同义密码子或者使用一些修饰核苷酸可以提高蛋白质的翻译效率,但是这种方法也有可能改变mrna的二级结构,影响动力学的同时影响蛋白的折叠。不同的蛋白的二级结构会影响效应t淋巴细胞表达表位肽的不同阅读框架的表达。所以的这些细节都有可能会影响mrna疫苗引发的免疫应答。
15.药用mrna技术的快速发展同时也对mrna大规模生产的工艺、产率、质量以及成本等提出了很大的挑战,特别是在mrna疫苗的超大规模生产过程当中,对于mrna自身的质量控制更是显得尤为重要。
[0016]5’
cap作为mrna的重要结构,是mrna稳定存在并进行蛋白质高效率表达的重要保证。现阶段通过体外转录方法合成的mrna主要通过帽子类似物和加帽酶的方法对mrna进行加帽,最终的mrna产品是否进行了正确的加帽并取得合格的加帽效率则成为了评价mrna产品的重要指标。并且对于不同的应用场景需要对mrna加帽的种类进行区分,也就是需要精确测定mrna的cap 0和cap 1的占比。现有加帽效率的测定方法是将mrna水解后测定m7g与a,u,c,g四种碱基的比例来比较出mrna的加帽效率,但是由于体外转录的工艺限制,mrna的纯度无法达到100%,所以基于此类方法不能够准确测定mrna的加帽效率。同时对于不同种类不同序列的mrna需要准确知道各碱基的个数才可以进行测量,所以方法并不具有通用性。
[0017]
此外,利用常规atp、gtp、ctp、utp并体外转录技术取得的mrna会引发机体的自有免疫反应,导致mrna的降解。修饰核苷酸如假尿苷、5甲基胞嘧啶的加入可以有效的抑制mrna自身的免疫原性,防止mrna在体内被降解,并可以延长mrna在细胞内的表达时间,提高表达效率。所以修饰核苷酸在整个mrna的占比也是评价mrna产品的重要指标,经过充分修饰的mrna才能够拥有较好的功能性。
[0018]
体外转录的mrna不可避免的会与空气中的氧气发生接触并导致mrna的氧化,这当中的氧化产物则以8氧鸟苷为主,8氧鸟苷会导致转录出错,影响蛋白的正确表达。能够准确的判断mrna氧化产物是评判mrna药物的一个重要标准。
[0019]
迄今为止,非常多的方法已经被开发用于mrna产品的检测,比如通过电泳来判断mrna的长度及纯度,通过pcr及测序技术判断mrna的序列正确性及poly a尾巴长度。通过hplc,gc/ms,lc/ms的方法对加帽效率,修饰核苷酸占比和氧化产物的检测也已经被开发出来。例如专利申请wo2017149139a1中所涉及的方法,然而该方法仅适用于mrna的纯度为100%时的加帽效率测定。受限于mrna的生产工艺,mrna纯度不可能达到100%,所以有大量截短的mrna仍然会被加帽,这样会导致mrna的加帽效率测定时会大于100%,所以该方法在原理上是不精确的。可见,现有的所有方法因无法精确测定待测样品中mrna的物质的量导致难以精确测定加帽效率。
技术实现要素:
[0020]
本发明所要解决的技术问题是为克服现有技术中缺乏加帽效率的检测方法以及现有方法依赖于mrna纯度的检测的缺陷,提供一种测定mrna平均分子量以及cap0/1、修饰核苷酸及氧化物的方法。
[0021]
现有技术中因mrna纯度检测并不准确,所以基于此类方法测定的加帽效率、修饰核苷酸占比及mrna氧化的结果并不准确。本方法创造性地利用毛细管电泳对mrna的纯度及链长分布进行精确的测定,并将mrna完全水解后对rna的例如,7-甲基-鸟苷,2-甲氧基-鸟苷,体外转录时添加的修饰核苷酸,8-氧-鸟苷等特征性化合物进行测定,无需预先测定或知道mrna的序列及各种不同碱基的含量,通过本发明登载的算法即可精确测得mrna的cap0、cap1,修饰核苷酸及氧化物比例。实现对mrna质量的精确评价。
[0022]
本发明主要通过以下技术方案解决上述技术问题。
[0023]
本发明的技术方案之一为:一种测定样品中mrna平均分子量的方法,其包括:
[0024]
(1)获得样品的毛细管电泳图;
[0025]
(2)根据公式(i)
[0026][0027]
计算得mrna平均分子量,其中,a为毛细管电泳中所检测到的最短mrna的碱基个数,b为最长碱基个数,f(n)=mrna所占比例,n=mrna总碱基个数。
[0028]
在本发明一实施方案中,将毛细管电泳图横坐标分为n等分并命名为a1、a2…an-1
、an,其中an=b,所述n≥5,优选地,n≥10;通过如下公式(ii)对所述公式(1)进行求解:
[0029][0030]
所述r为区间(a
n-1
,an)内的峰面积占比。
[0031]
其中,关于n的数值做如下说明:n无限趋近于正无穷时计算结果无限趋近真实值,因此,在本发明中若取样更多,则结果相对更为精确,但是n取值过大会造成计算十分繁琐,影响效率。本发明中,n可为5~100或5~50或5~20或10~100或10~50或5~20的任何正整数,例如5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20、30、40、50等。
[0032]
步骤(1)中所述毛细管电泳可按照本领域常规进行,较佳地使用small rna reagent kit以及gx touch
tm
nucleic acid analyzer进行毛细管电泳,以获得样品的毛细管电泳图。
[0033]
在本发明一较佳实施方案中,所述样品在进行毛细管电泳前还包括除去游离核苷酸的步骤;所述除去游离核苷酸例如采用licl沉淀法,离子交换树脂层析,制备级液相色谱或商业用试剂盒进行。
[0034]
本发明的技术方案之二为:一种测定样品中mrna的物质的量的方法,其包括如下步骤:
[0035]
1)测定样品中mrna中的质量浓度,计算样品中mrna的质量;
[0036]
2)根据如技术方案之一所述的方法,测定样品中mrna平均分子量;
[0037]
3)根据步骤1)所述的mrna的质量和步骤2)中所得样品中mrna平均分子量得出样品中mrna的物质的量。
[0038]
步骤1)中,mrna中的质量浓度可通过本领域常规,较佳地使用超微量分光光度计测定样品中mrna的质量浓度;所述超微量分光光度计例如为thermo scientific nanodrop one或者mettler toledo uv5 nano。
[0039]
步骤3)中,所提及的“物质的量”可通过本领域常规方法进行计算,即:mrna的物质的量=mrna的质量/mrna平均分子量。
[0040]
本发明的技术方案之三为:一种mrna中核苷酸含量的测定方法,所述测定方法包括:
[0041]
(a)通过如技术方案之二所述的方法获得所述mrna的物质的量;
[0042]
(b)测定不同的所述核苷酸的物质的量;
[0043]
(c)根据步骤(a)中不同的所述核苷酸的物质的量和步骤(b)中所述mrna的物质的量,计算不同的所述核苷酸在mrna中的含量;
[0044]
以上步骤(a)和(b)没有顺序要求,能够分别、同时进行或者先后进行。
[0045]
以上步骤(b)中,优选通过hplc测定水解的mrna中不同核苷酸的含量,根据其相对分子质量计算得到不同核苷酸的物质的量。所述水解包括如下步骤:
[0046]
i.将待水解mrna与核酸内切酶p1混合,并加入缓冲液a后42℃孵育;所述孵育的时间例如为2小时;所述待水解mrna、核酸内切酶p1与缓冲液a的体积比例如为100:10:33;
[0047]
ii.向步骤i所得孵育产物中添加磷酸二酯酶i和碱性磷酸酶,并加入缓冲液b,于37℃孵育;所述孵育的时间例如为2小时,所述磷酸二酯酶i、碱性磷酸酶以及缓冲液b的体积比例如为3.3:3.3:10.6。
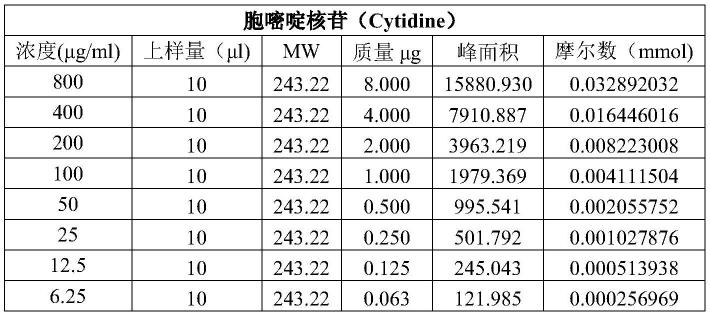
[0048]
本发明中所述的hplc测定可为本领域常规,较佳地包括:
[0049]
i)使用不同浓度的特定核苷酸的标准品制作浓度与峰面积的标准曲线;
[0050]
ii)获得不同的所述核苷酸的峰面积后,对应所述标准曲线计算得所述核苷酸的浓度。
[0051]
以上所述hplc的参数例如为:
[0052]
柱温25℃,流速0.85ml/min,检测波长254nm,流动相a为5mm醋酸铵水溶液,流动相b为40%乙腈水溶液,梯度100%a—20%b,50min。
[0053]
所测定的核苷酸的种类可为常规的、构成mrna的核苷,例如腺嘌呤核苷、尿嘧啶核苷、胞嘧啶核苷以及鸟嘌呤核苷;也可为其相应的修饰核苷酸或者氧化后核苷酸以及其他常见的非常规核苷。
[0054]
本发明中所述修饰核苷酸优选包括7-甲基-鸟嘌呤核苷、2
’‑
氧甲基-鸟嘌呤核苷、假尿嘧啶核苷、1-甲基-假尿嘧啶核苷、5-甲基-胞嘧啶核苷、4-乙酰基-胞嘧啶核苷以及6-甲基-腺嘌呤核苷中的一种或多种。
[0055]
在本发明一较佳实施方案中:
[0056]
a.所述7-甲基鸟嘌呤核苷的占比通过如下公式获得:
[0057]
cap0%=7-甲基鸟嘌呤核苷物质的量/mrna物质的量
×
100%;
[0058]
b.所述2-氧甲基-鸟嘌呤核苷的占比通过如下公式获得:
[0059]
cap1%=2-氧甲基-鸟嘌呤核苷物质的量/mrna物质的量
×
100%
×
cap0%;
[0060]
c.所述修饰核苷酸的占比通过如下公式计算:
[0061]
修饰核苷酸%=修饰核苷酸的物质的量/(修饰核苷酸的物质的量 修饰前核苷酸的物质的量)
×
100%;
[0062]
d.所述氧化后核苷酸的占比通过如下公式计算:
[0063]
氧化后核苷酸%=氧化后核苷酸的物质的量/(氧化后核苷酸的物质的量 氧化前核苷酸的物质的量)
×
100%。
[0064]
所述修饰核苷酸可为本领域常规,较佳地包括:1-甲基-假尿嘧啶核苷、5-甲基-胞嘧啶核苷、4-乙酰基-胞嘧啶核苷以及6-甲基-腺嘌呤核苷中的一种或多种。
[0065]
所述氧化后核苷酸也可为本领域常规,例如8-氧-鸟嘌呤核苷。
[0066]
在符合本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
[0067]
本发明所用试剂和原料均市售可得。
[0068]
本发明的积极进步效果在于:
[0069]
本发明通过一种新的测定及计算方法对mrna制备中的cap 0、cap 1、修饰核苷酸及氧化产物占比进行精确的测定,本发明对于药用及科研用的mrna的质量控制提供了非常精确的检测方法。
附图说明
[0070]
图1为毛细管电泳图谱。
[0071]
图2为水解mrna图谱。
[0072]
图3为胞嘧啶核苷的标准曲线。
[0073]
图4为尿嘧啶核苷的标准曲线。
[0074]
图5为鸟嘌呤核苷的标准曲线。
[0075]
图6为7-甲基-鸟嘌呤核苷的标准曲线。
[0076]
图7为2-氧甲基-鸟嘌呤核苷的标准曲线。
[0077]
图8为假尿嘧啶核苷的标准曲线。
[0078]
图9为8-氧-鸟嘌呤核苷酸的标准曲线。
具体实施方式
[0079]
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
[0080]
实施例1游离核苷酸的去除
[0081]
mrna的链长分布测定可以使用labchip gxii touch ht(perkinelmer),2100electrophoresis bioanalyzer instrument(agilent),qsep100(bioptic)等仪器完成,所有仪器测定都可以得到相同的测定结果。本文中提到的mrna链长分布都以labchip gxii touch ht的测定结果为例。mrna可采用licl沉淀法,离子交换树脂层析,制备级液相色谱或商业用试剂盒等方法进行纯化,完全除去游离核苷酸以防止对各项指标测定产生干扰。
[0082]
实施例2mrna完全水解为单核苷并利用hplc对各水解后的组分进行鉴定
[0083]
1)实验试剂与设备:
[0084]
核酸内切酶p1(endonuclease p1)、磷酸二酯酶i(phosphodiesterase i)以及碱性磷酸酶(shrimp alkaline phosphatase)购于sigma aldrich。
[0085]
氯化锌、甘油、醋酸钠、醋酸铵以及醋酸镁均采购自国药试剂集团。超纯水则都取自milli-q纯水机。
[0086]
尿嘧啶核苷(uridine)、胞嘧啶核苷(cytidine)以及鸟嘌呤核苷(guanosine)均购于merck;假尿嘧啶核苷(pseudouridine)、7-甲基鸟嘌呤核苷(7-methyl guanosine)、8-氧-鸟嘌呤核苷(8-oxo-guanosine)以及2
’‑
氧甲基-鸟嘌呤核苷(2
’‑
o-methylguanosine,简称2
’‑
ome-guanosine)购于tci。
[0087]
高效液相色谱仪(1260infinity ii bioinert lc)购置于安捷伦科技,高效液相色谱柱(xbridge beh c18 column,5μm,4.6mm x 250mm,1/pk)购置于waters,台式离心机(st8)与恒温振荡金属浴(88880028)购置于thermo,分析天平(xs205du)购置于multiparameter,微量移液器购置于rainin。
[0088]
2)实验方法与操作:
[0089]
在贮存液(110mm tris-hcl缓冲液、100mm氯化钠、15mm氯化镁、50%甘油,ph 8.9)中加入7g/l磷酸二酯酶i(phosphodiesterase i),配制为磷酸二酯酶i(phosphodiesterase i)贮存液,储存于-20℃。
[0090]
核酸内切酶p1(endonuclease p1)配制为1g/l的贮存液(20mm醋酸钾、5mm氯化锌、50mm氯化钠以及50%甘油)储存于-20℃。
[0091]
缓冲液a(141.43mm醋酸铵、9.43mm氯化锌,ph 5.3)。
[0092]
缓冲液b(115.00mm tris-hcl、11.50mm醋酸镁,ph 8.3)。
[0093]
将100μl待测mrna(3.018mg/ml)样品加入2μl稀释10倍的核酸内切酶p1(endonuclease p1),并加入33μl缓冲液a后42℃孵育2小时,加入3.3μl磷酸二酯酶i(phosphodiesterase i)贮存液和碱性磷酸酶(shrimp alkaline phosphatase)(0.1u/μl)补加缓冲液b10.6μl并继续37℃孵育2小时。mrna降解完成后17000g离心10min,取出上清液(体积153.3μl,mrna降解物1.969mg/ml)检测备用。将hplc调节至如下参数:柱温25℃,流速0.85ml/min,检测波长254nm,流动相a为5mm醋酸铵水溶液,流动相b为40%乙腈水溶液,梯
度100%a—20%b,50min。可取得水解mrna图谱如图2所示。
[0094]
通过比对不同核苷酸的保留时间并积分可得到核苷酸峰面积:
[0095]
假尿嘧啶核苷(pseudouridine):4263.484,尿嘧啶核苷(uridine):12.689,7-甲基鸟嘌呤核苷(7-methyl guanosine):18.998,2
’‑
氧甲基-鸟嘌呤核苷(2
’‑
ome-guanosine):31.655,8-氧代-鸟嘌呤核苷(8-oxo-guanosine):0。
[0096]
将尿嘧啶核苷、胞嘧啶核苷、鸟嘌呤核苷、假尿嘧啶核苷(pseudouridine)、7-甲基鸟嘌呤核苷(7-methyl guanosine)、8-氧-鸟嘌呤核苷(8-oxo-guanosine)以及2
’‑
氧甲基-鸟嘌呤核苷(2
’‑
ome-guanosine)等多种核苷配制成梯度浓度水溶液利用hplc进行测量,每个样品进样量10μl,记录不同核苷标准品的保留时间,并利用归一法积分计算不同核苷在不同浓度下的峰面积,并由此依据核苷的梯度浓度绘制核苷的标准曲线。
[0097]
根据如下表格1~7绘制得如图3~图9所示的各核苷标准曲线。
[0098]
表1
[0099][0100]
表2
[0101][0102]
表3
[0103][0104]
表4
[0105][0106][0107]
表5
[0108][0109]
表6
[0110][0111]
表7
[0112][0113]
根据不同核苷的标准曲线和峰面积可以计算得出单位进样量(10μl)中不同核苷的浓度如下所示:假尿嘧啶核苷(pseudouridine):8.64e-03μmol,尿嘧啶核苷(uridine):2.97e-05μmol,7-甲基-鸟嘌呤核苷(7-methyl guanosine):2.96e-05μmol,2
’‑
氧甲基-鸟嘌呤核苷(2
’‑
ome-guanosine):3.02e-05μmol,8-氧-鸟嘌呤核苷(8-oxo-guanosine):0(未检出)。
[0114]
3、利用本发明的算法计算mrna的各项指标。
[0115]
1)精确标定样品中mrna的质量浓度。
[0116]
首先利用nanodrop精确测定样品中mrna的质量浓度,nanodrop可以选用thermo scientific nanodrop one,mettler toledo uv5 nano等类似设备。
[0117]
2)精确测定样品mrna的平均分子质量。
[0118]
考虑到依据现有的体外转录法进行mrna大规模合成的成品mrna很难达到100%纯度,所以仅仅使用经验公式:
[0119]
mw(rna)=(a
×
329.2) (u
×
306.2) (c
×
305.2) (g
×
345.2) 159
[0120]
无法进行准确的mrna的分子量进行计算。
[0121]
本发明使用利用毛细管电泳图谱与创新性算法相结合,无需知晓mrna碱基序列及各碱基个数的前提下就可以对样品mrna序列进行精确标定从而计算cap0、cap1、修饰核苷酸及氧化物的含量。
[0122]
当mrna的纯度为100%时,mrna的分子量的计算方法如下:
[0123]
mrna分子量可以精确计算为:
[0124]
mw=(a
×
329.2) (u
×
306.2) (c
×
305.2) (g
×
345.2) 159
[0125]
当mrna长度大于1000nt后mrna分子量可以近似为:
[0126]
mw=n
×
320.5 159(n=mrna总碱基个数)
[0127]
但是基于现有mrna制备工艺,100%纯度是不能达到的。如果mrna的纯度无法达到100%,则需要利用mrna的毛细管电泳图谱进行测定。
[0128]
将毛细管电泳图谱曲线定义为函数:
[0129]
r=f(n),n=mrna总碱基个数,r=mrna所占比例。
[0130]
样品mrna的平均分子量则可以由此计算得出:
[0131][0132]
a为毛细管电泳图中可检测到的最短mrna碱基个数,b为毛细管电泳图中可检测到的最长碱基个数。
[0133]
函数f(n)的求解为“求平面坐标系中任意曲线的函数表达式”,求解此函数可以借用高等数学的“多项式拟合方程”,或者借用python及matlab进行求解。
[0134]
此方程可以借助wolframalpha进行求解(https://www.wolframalpha.com/),或者采用本发明提供的近似算法:
[0135]
将毛细管电泳图横坐标分为n(n≥10,本实施例中n=11)等分即a1,a2…an-1
,an,其中an=b(本实施例中b=2946)。其中r为区间(a
n-1
,an)内的峰面积占比。
[0136]
mrna平均分子量可以近似计算为:
[0137][0138]
表8
[0139][0140]
如表8和图1所示,将待测mrna毛细管电泳图谱进行分割后,得出不同长度区间的mrna的分配比例。然后代入以上公式可以得出mrna的平均分子量及单位进样量(10μl)中mrna的摩尔数,结果见下表9。
[0141]
表9
[0142]
[0143][0144]
3)cap0及cap1占比计算
[0145]
mrna的cap结构如下所示:
[0146][0147]
其中7-甲基-鸟嘌呤核苷(7-methyl guanosine)是cap0结构的标志物,每个完整的mrna有且仅有一个7-甲基-鸟嘌呤核苷(7-methyl guanosine):
[0148][0149]
因此可以通过精确测定7-甲基-鸟嘌呤核苷(7-methyl guanosine)的含量,样品mrna的cap0占比可以计算为:
[0150]
cap0%=7-甲基-鸟嘌呤核苷(7-methyl guanosine)物质的量/mrna物质的量
×
100%。
[0151]
根据实验测得结果cap0%=2.96e-05μmol/3.32e-05μmol=89%。
[0152]
7-甲基-鸟嘌呤核苷(7-methyl guanosine)与2-氧甲基-鸟嘌呤核苷(2-ome-guanosine)通过三磷酸键相连的复合结构是mrna的cap1结构的标志物,每个完整的mrna有且仅有一个7-甲基-鸟嘌呤核苷(7-methyl guanosine)与2
’‑
氧甲基-鸟嘌呤核苷(2
’‑
ome-guanosine)的复合结构。因此通过测定标志物2
’‑
氧甲基-鸟嘌呤核苷(2
’‑
ome-guanosine)的物质的量即可得到cap1的占比:
[0153][0154]
cap1%=2
’‑
氧甲基-鸟嘌呤核苷(2
’‑
ome-guanosine)物质的量/mrna物质的量
×
100%
×
cap0%。
[0155]
根据实验结果测得cap1%=3.02e-05μmol/3.32e-05μmol
×
89%=81%。
[0156]
4)修饰核苷酸占比测定
[0157]
为提高药用mrna的表达效率并延长表达时间,在体外转录的过程中往往需要加入假尿嘧啶核苷(pseudouridine)、1-甲基-假尿嘧啶核苷(1-methyl-pseudouridine)、5-甲基-胞嘧啶核苷(5-methyl-cytidine)、4-乙酰-胞嘧啶核苷(4-acetyl-cytidine)以及6-甲基-腺嘌呤核苷(6-methyl-adenesine)等修饰核苷酸。
[0158][0159]
为测定相应修饰核苷酸在mrna样品中的占比可以利用如下公式进行计算。
[0160]
假尿嘧啶核苷(pseudouridine)%=假尿嘧啶核苷(pseudouridine)物质的量/(尿嘧啶核苷(uridine)物质的量 假尿嘧啶核苷(pseudouridine)物质的量)
×
100%。
[0161]
本实验可以精确测得:假尿嘧啶核苷(pseudouridine)%=8.64nmol/(8.64nmol 2.97e-02nmol)
×
100%=99.7%。
[0162]
1-甲基-假尿嘧啶核苷(1-methyl-pseudouridine)%=1-甲基-假尿嘧啶核苷(1-methyl-pseudouridine)物质的量/(1-甲基-假尿嘧啶核苷(1-methyl-pseudouridine)物质的量 尿嘧啶核苷(uridine)物质的量)
×
100%。
[0163]
5-甲基-胞嘧啶核苷(5-methyl-cytidine)%=5-甲基-胞嘧啶核苷(5-methyl-cytidine)物质的量/(5-甲基-胞嘧啶核苷(5-methyl-cytidine)物质的量 胞嘧啶核苷(cytidine)物质的量)
×
100%。
[0164]
4-乙酰基胞嘧啶核苷(4-acetyl-cytidine)%=4-acetyl-cytidine物质的量/(4-acetyl-cytidine物质的量 cytidine物质的量)
×
100%。
[0165]
6-甲基-腺嘌呤核苷(6-methyl-adenesine)%=6-甲基-腺嘌呤核苷(6-methyl-adenesine)物质的量/(6-甲基-腺嘌呤核苷(6-methyl-adenesine)物质的量 腺嘌呤核苷(adenesine)物质的量)
×
100%。
[0166]
其它任意修饰核苷酸占比都可采用同样的方法进行精确计算。
[0167]
5)氧化产物占比测定
[0168]
mrna长期与空气接触会不可避免的发生氧化。其氧化反应主要表现在鸟嘌呤核苷(guanosine)转化为8-氧-鸟嘌呤核苷(8-oxo-guanosine)。
[0169][0170]
对8-氧-鸟嘌呤核苷(8-oxo-guanosine)的浓度进行精确测定可以直接得出mrna的氧化产物含量。
[0171]
oxidation%=8-氧-鸟嘌呤核苷(8-oxo-guanosine)物质的量/(8-氧-鸟嘌呤核苷(8-oxo-guanosine)物质的量 鸟嘌呤核苷(guanosine)物质的量)
×
100%。
[0172]
本实验中mrna氧化产物未检出。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。