1.本发明属于医药环境化工领域,特别是涉及一种高品质9β,11β-环氧-孕甾-1,4,16-三烯-3,20-二酮-21-醋酸酯的制备方法。
背景技术:
2.9β,11β-环氧-孕甾-1,4,16-三烯-3,20-二酮-21-醋酸酯是一种重要的甾体药物中间体,可用于合成地塞米松、曲安奈德、丙酸氟替卡松和氟米松等重要甾体激素药物。9β,11β-环氧-孕甾-1,4,16-三烯-3,20-二酮-21-醋酸酯呈类白色或淡黄色结晶性粉末,其结构式如下:
[0003][0004]
9β,11β-环氧-孕甾-1,4,16-三烯-3,20-二酮-21-醋酸酯的合成是以3tr为起始原料,经溴羟化反应和环氧闭环反应得到,具体合成路线如下:
[0005][0006]
在溴羟化反应中,原料3tr残留在0.2%以下。但由于在闭环反应过程中,会有副反应产生,导致杂质3tr在0.5%~1.0%,副反应方程式如下:
[0007]
[0008]
在原合成工艺中,环氧闭环反应完毕后,向反应液中加大量水析料,采用该方法得到的9β,11β-环氧-孕甾-1,4,16-三烯-3,20-二酮-21-醋酸酯产品中,杂质3tr在0.5%~1.0%,一般精制方法难以脱除。在下游产品中,3tr衍生杂质偏大,导致下游产品不合格,需增加精制工序。
[0009]
为满足下游产品市场需求,需进一步提高9β,11β-环氧-孕甾-1,4,16-三烯-3,20-二酮-21-醋酸酯产品质量。
技术实现要素:
[0010]
本发明所要解决的技术问题是提供一种高品质甾体药物中间体的制备方法,以解决上述技术问题。
[0011]
本发明公开了一一种高品质甾体药物中间体的制备方法,以其合成反应液为起始物料。合成反应液是以醋酸四烯物(3tr)为起始物料,经溴羟化反应和环氧闭环反应得到。其中,溴羟化反应:以丙酮为溶剂、二溴海因为溴羟化试剂,在高氯酸催化作用下,3tr转化为溴羟物;环氧闭环反应:向溴羟化反应液中加入碳酸钠水溶液,在碱性作用下,得到9β,11β-环氧-孕甾-1,4,16-三烯-3,20-二酮-21-醋酸酯合成反应液。
[0012]
该高品质甾体药物中间体的制备方法,包括以下步骤:
[0013]
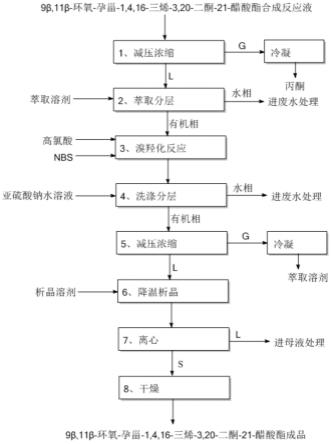
步骤(1)、减压浓缩:将合成反应液减压浓缩,气相冷凝后得到丙酮,固液混合物料进入下一步;
[0014]
步骤(2)、萃取分层:向步骤(1)得到的固液混合物料中加入萃取溶剂,混合1h,静置1h,分层,水相进废液处理,有机相进入下一步;
[0015]
步骤(3)、溴羟化反应:向步骤(2)得到的有机相中加入高氯酸和nbs,于20~30℃混合反应1~3h,反应完毕,进入下一步;
[0016]
步骤(4)、亚硫酸钠水溶液洗涤分层:向步骤(3)的反应液中加入质量浓度为10%的亚硫酸钠水溶液,混合1h,静置1h,分层,水相进废液处理,有机相进入下一步;
[0017]
步骤(5)、减压浓缩:将步骤(4)得到的有机相减压浓缩,气相冷凝后得到萃取溶剂,向浓缩后的料液中加入析晶溶剂,固液混合物料进入下一步;
[0018]
步骤(6)、降温析晶:将步骤(5)得到的固液混合物料降温至-15~0℃,保温1~5h,进入下一步;
[0019]
步骤(7)、离心:将步骤(6)中的物料离心,母液进入废液处理,固体物料进入下一步;
[0020]
步骤(8)、将步骤(7)中的固体物料转入真空干燥设备中,于45~60℃干燥,得高品质9β,11β-环氧-孕甾-1,4,16-三烯-3,20-二酮-21-醋酸酯成品。
[0021]
可选地,步骤(2)中,所述萃取溶剂为二氯甲烷、氯仿和甲苯中至少一种;
[0022]
所述萃取溶剂与所述3tr的质量比为1~5:1。
[0023]
可选地,步骤(3)中,所述高氯酸与所述3tr的质量比为0.01~0.03:1;
[0024]
所述nbs与所述3tr的质量比为0.02~0.05:1。
[0025]
可选地,步骤(4)中,所述质量浓度为10%的亚硫酸钠水溶液与所述3tr的质量比为1~5:1。
[0026]
可选地,步骤(5)中,所述析晶溶剂为甲醇、异丙醇和乙酸乙酯中至少一种;
[0027]
所述析晶溶剂与所述3tr的质量比为1~5:1。
[0028]
与现有技术相比,本发明包括以下优点:
[0029]
(1)、本发明采用减压浓缩技术实现丙酮的回收套用,采用萃取分层技术实现体系中醋酸钠和氯化钠及大部分二甲基海因(二溴海因降解产物)等物质与产品分离,相比于原工艺,减少了废水的产生;
[0030]
(2)、本发明采用减压浓缩和萃取分层技术实现物料体系溶媒置换,有利于产品中的杂质3tr发生溴羟化反应,将3tr转化为溴羟物,溴羟物比3tr更易于从9β,11β-环氧-孕甾-1,4,16-三烯-3,20-二酮-21-醋酸酯中分离;
[0031]
(3)、本发明选择活性较高的nbs作为溴羟化试剂,可以实现低浓度的杂质3tr顺利转化为溴羟物;
[0032]
(4)、本发明充分利用溴羟物与9β,11β-环氧-孕甾-1,4,16-三烯-3,20-二酮-21-醋酸酯的溶解度差异,选择甲醇、异丙醇和乙酸乙酯中至少一种作为析晶溶剂,可以有效地脱除产品中的杂质3tr;
[0033]
(5)、本发明的工艺合理,产品质量好,产品收率高的特点,易于实现大规模工业化生产。该工艺方法得到的成品纯度在99%以上,特定杂质3tr在0.1%以下,摩尔收率在95%以上。
附图说明
[0034]
图1是本发明的工艺流程图。
具体实施方式
[0035]
为使本发明的上述目的、特征和优点能够更加明显易懂,下面结合附图和具体实施方式对本发明作进一步详细的说明。
[0036]
下面通过实施例对本发明所述方法的实现流程进行详细说明。
[0037]
合成反应液是以醋酸四烯物(3tr)为起始物料,经溴羟化反应和环氧闭环反应得到。其中,溴羟化反应:以丙酮为溶剂、二溴海因为溴羟化试剂,在高氯酸催化作用下,3tr转化为溴羟物;环氧闭环反应:向溴羟化反应液中加入碳酸钠水溶液,在碱性作用下,得到9β,11β-环氧-孕甾-1,4,16-三烯-3,20-二酮-21-醋酸酯合成反应液。
[0038]
实施例1一种高品质甾体药物中间体的制备方法,以其合成反应液为起始物料,步骤如下:
[0039]
步骤(1)、减压浓缩:将合成反应液减压浓缩,气相冷凝后得到丙酮,固液混合物料进入下一步;
[0040]
步骤(2)、萃取分层:向步骤(1)得到的固液混合物料中加入100ml二氯甲烷,混合1h,静置1h,分层,水相进废液处理,有机相进入下一步;
[0041]
步骤(3)、溴羟化反应:向步骤(2)得到的有机相中加入2g高氯酸和10g nbs,于30℃混合反应1h,反应完毕,进入下一步;
[0042]
步骤(4)、亚硫酸钠水溶液洗涤分层:向步骤(3)的反应液中加入1000g质量浓度为10%的亚硫酸钠水溶液,混合1h,静置1h,分层,水相进废液处理,有机相进入下一步;
[0043]
步骤(5)、减压浓缩:将步骤(4)得到的有机相减压浓缩,气相冷凝后得到二氯甲
烷,向浓缩后的料液中加入200g甲醇,固液混合物料进入下一步;
[0044]
步骤(6)、降温析晶:将步骤(5)得到的固液混合物料降温至0℃,保温5h,进入下一步;
[0045]
步骤(7)、离心:将步骤(6)中的物料离心,母液进入废液处理,固体物料进入下一步;
[0046]
步骤(8)、将步骤(7)中的固体物料转入真空干燥设备中,于45℃干燥,得高品质9β,11β-环氧-孕甾-1,4,16-三烯-3,20-二酮-21-醋酸酯成品198.4g,摩尔收率95.1%,纯度99.2%,杂质3tr大小为0.03%。
[0047]
实施例2一种高品质甾体药物中间体的制备方法,以其合成反应液为起始物料,步骤如下:
[0048]
步骤(1)、减压浓缩:将合成反应液减压浓缩,气相冷凝后得到丙酮,固液混合物料进入下一步;
[0049]
步骤(2)、萃取分层:向步骤(1)得到的固液混合物料中加入300ml氯仿,混合1h,静置1h,分层,水相进废液处理,有机相进入下一步;
[0050]
步骤(3)、溴羟化反应:向步骤(2)得到的有机相中加入2g高氯酸和3g nbs,于25℃混合反应2h,反应完毕,进入下一步;
[0051]
步骤(4)、亚硫酸钠水溶液洗涤分层:向步骤(3)的反应液中加入300g质量浓度为10%的亚硫酸钠水溶液,混合1h,静置1h,分层,水相进废液处理,有机相进入下一步;
[0052]
步骤(5)、减压浓缩:将步骤(4)得到的有机相减压浓缩,气相冷凝后得到氯仿,向浓缩后的料液中加入300g异丙醇,固液混合物料进入下一步;
[0053]
步骤(6)、降温析晶:将步骤(5)得到的固液混合物料降温至-10℃,保温3h,进入下一步;
[0054]
步骤(7)、离心:将步骤(6)中的物料离心,母液进入废液处理,固体物料进入下一步;
[0055]
步骤(8)、将步骤(7)中的固体物料转入真空干燥设备中,于50℃干燥,得高品质9β,11β-环氧-孕甾-1,4,16-三烯-3,20-二酮-21-醋酸酯成品99.5g,摩尔收率95.4%,纯度99.3%,杂质3tr大小为0.05%。
[0056]
实施例3一种高品质甾体药物中间体的制备方法,以其合成反应液为起始物料,步骤如下:
[0057]
步骤(1)、减压浓缩:将合成反应液减压浓缩,气相冷凝后得到丙酮,固液混合物料进入下一步;
[0058]
步骤(2)、萃取分层:向步骤(1)得到的固液混合物料中加入750g甲苯,混合1h,静置1h,分层,水相进废液处理,有机相进入下一步;
[0059]
步骤(3)、溴羟化反应:向步骤(2)得到的有机相中加入4.5g高氯酸和2g nbs,于20℃混合反应3h,反应完毕,进入下一步;
[0060]
步骤(4)、亚硫酸钠水溶液洗涤分层:向步骤(3)的反应液中加入150g质量浓度为10%的亚硫酸钠水溶液,混合1h,静置1h,分层,水相进废液处理,有机相进入下一步;
[0061]
步骤(5)、减压浓缩:将步骤(4)得到的有机相减压浓缩,气相冷凝后得到甲苯,向浓缩后的料液中加入750g乙酸乙酯,固液混合物料进入下一步;
[0062]
步骤(6)、降温析晶:将步骤(5)得到的固液混合物料降温至-15℃,保温1h,进入下一步;
[0063]
步骤(7)、离心:将步骤(6)中的物料离心,母液进入废液处理,固体物料进入下一步;
[0064]
步骤(8)、将步骤(7)中的固体物料转入真空干燥设备中,于60℃干燥,得高品质9β,11β-环氧-孕甾-1,4,16-三烯-3,20-二酮-21-醋酸酯成品143g,摩尔收率95.3%,纯度99.1%,杂质3tr大小为0.07%。
[0065]
以上对本发明所提供的一种高品质甾体药物中间体的制备方法进行了详细介绍,本文中应用了具体个例对本发明的原理及实施方式进行了阐述,以上实施例的说明只是用于帮助理解本发明的方法及其核心思想;同时,对于本领域的一般技术人员,依据本发明的思想,在具体实施方式及应用范围上均会有改变之处,综上所述,本说明书内容不应理解为对本发明的限制。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。
