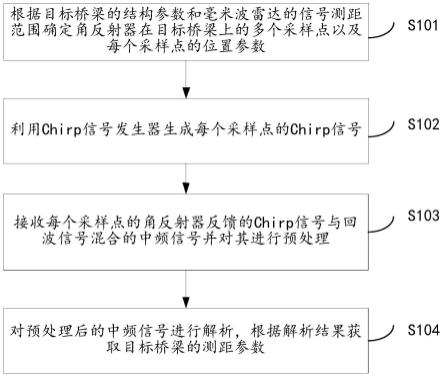
1.本发明属于中药质量控制技术领域,具体涉及以甘草对照药材为基准物质,建立一套不依赖于对照品,较为全面控制药材质量的方法,该方法稳定性、精密度良好,可以弥补目前甘草药材质量控制的不足,能够为甘草药材质量提升提供新思路。
背景技术:
2.甘草药材为豆科植物甘草glycyrrhiza uralensis fisch.的干燥根及根茎,在我国西北、东北、华北地区均有分布,主产于新疆、甘肃、河北等地。其性平味甘,在临床上常作为清热解毒,祛痰止咳之要药,常用于调和诸药,然而不同产地甘草药材质量参差不齐,影响其在临床上的合理应用,因此甘草药材的质量控制十分重要。
3.随着现代中药分析手段的进步,甘草药材质量控制方法主要包括指纹图谱法、高效毛细管电泳法、微乳薄层色谱法等,以上方法具有灵敏度高、重复性好的优点,能够为控制甘草药材提供量化数据,但存在着单一指标不能反映药材整体特征、多指标含量测定依赖于多种对照品且某些指标成分不能反映药效、指纹图谱只能模糊地评价药材相似性不能清晰地判断供试品真伪优劣的不足。
技术实现要素:
4.针对上述问题,本发明提供一种甘草药材质-量双标可视化质量控制技术。以甘草对照药材为定性、定量的基准物质,采用高效液相色谱结合q-tof-ms技术,建立一套不依赖于对照品,较为全面科学地控制药材质量的方法。
5.为了实现上述目的,本发明提供了如下技术方案。
6.本发明提供了一种甘草药材质量控制标准的建立方法,其特征在于,所述方法包括以下步骤:步骤1 基于对照药材的甘草药材定性研究;步骤2 基于内标物质的特征峰化学成分相对定量研究;步骤3 方法学考察。
7.进一步地,所述步骤1的具体步骤包括:(1)溶液的制备:取甘草对照药材粉末,精密称定,置容量瓶中,70%乙醇稀释至刻度,称定重量,超声(功率200 w,频率40 khz)30 min,放冷,再称定重量,用70%乙醇补足失重,摇匀,过0.22 μm微孔滤膜过滤,取续滤液,即得;(2)色谱条件与系统适用性试验:采用十八烷基硅烷键合硅胶为填充剂的agilent sb-c
18
色谱柱,色谱柱规格为4.6
×
100 mm,2.7-micron;以0.1%甲酸水为流动相a,以乙腈为流动相b,进行梯度洗脱;流速0.8 ml/min;柱温30℃;dad检测器在波长237 nm下进行检测;进样量5 μl;(3)特征图谱的建立:每份样品平行2次,按色谱条件依次进样检测,将237 nm波长下的图谱数据由分析检测仪器中导出获得10批供试药材的hplc叠加图谱;10批供试药材使
用中位数进行自动匹配,加以多点校正,生成共有模式r,与对照药材特征图谱进行比对;(4)相似度评价:以特征峰的相似度明确甘草药材真伪;供试药材与对照药材的相似度均大于0.96;(5)特征峰化学成分解析:采用q-tof-ms技术,通过分析化合物的保留时间、一级离子质荷比以及二级离子碎片信息,并与相关文献报道信息进行匹配,共鉴定出6个化学成分;(6)中药材(饮片)可视化的质-量双标质量控制方法程序:利用visual basic编程语言设计包含1个oel控件和4个command控件的适用于甘草药材(饮片)可视化的质-量双标质量控制程序。
8.更进一步地,所述(5)中鉴定出6个化学成分分别为:(3)号峰为芹糖甘草苷、(4)号峰为甘草苷、(6)号峰为芹糖异甘草苷、(8)号峰为异甘草苷、(12)号峰为异甘草素和(13)号峰为甘草酸。
9.进一步地,所述步骤2的具体步骤包括:以步骤1中确定的(13)号峰“甘草酸”作为内标物质,通过内标物质化学成分准确定量,计算10批供试药材特征峰化学成分的相对含量;取“中位数-标准差”作为特征峰化学成分相对于内标物质化学成分的相对含量下限。
10.进一步地,所述步骤2的特征峰均为甘草发挥其功效及药理作用的特征活性化学成分。
11.进一步地,所述步骤3的具体步骤包括:(1)精密度实验:精密吸取同一供试品溶液,按色谱条件测定,连续进样6次,测定各色谱峰相对保留时间、相对峰面积,计算相对标准偏差;各色谱峰相对保留时间、相对峰面积rsd值均小于1.0%,表明仪器精密度良好;(2)稳定性试验:精密吸取同一供试品溶液,按色谱条件,分别在0、2、4、8、12、24 h进样6次检测,测定各色谱峰相对保留时间、相对峰面积,计算相对标准偏差;各色谱峰相对保留时间、相对峰面积rsd值均小于1.2%,表明供试品溶液在24 h内稳定;(3)重复性实验:按供试品溶液制备方法制备6份供试样品,按色谱条件,分别进样检测,测定各色谱峰相对保留时间、相对峰面积,计算相对标准偏差;各色谱峰相对保留时间、相对峰面积rsd值均小于2.1 %,表明方法重复性良好;(4)线性关系考察:配制“甘草酸”的6个质量浓度溶液,按“2.1.2”项下色谱条件,分别进样检测,以质量(x)为横坐标,峰面积(y)为纵坐标,绘制标准曲线并进行线性回归,得到线性回归方程(y=3161.8x 30.754)、相关系数(r=0.999 6)。结果表明“甘草酸”在0.0550-0.3300 μg范围内线性关系良好。
12.进一步地,所述方法通过甘草药材特征图谱中的特征峰作为甘草药材的定性标准,能够明确鉴定药材的真伪。
13.进一步地,所述方法通过建立的特征峰相对含量的计算方法确定的各特征峰相对含量的下限,能够区分甘草的优劣。
14.进一步地,所述方法通过visual basic编程语言设计的可视化的质-量双标质量控制程序适用于甘草药材(饮片)。
15.与现有技术相比本发明的有益效果。
16.(1)本发明首次公开甘草药材质-量双标控制方法,可以弥补目前甘草药材质量控制方法中单一指标不能反映药材整体特征、多指标含量测定依赖于多种对照品且某些指标成分不能反映药效、指纹图谱只能模糊地评价药材相似性不能清晰地判断供试品真伪优劣的不足。
17.(2)本发明同一特征图谱不能适用于不同产地的甘草成分检测,绘制多个不同产地甘草药材的特征图谱,构建了更加全面的药材特征图谱。本文采用质谱技术指认出的特征峰化学成分指认出的特征峰化学成分主要为黄酮类和三萜皂苷类,其中黄酮类化合物包括芹糖甘草苷、甘草苷、芹糖异甘草苷、异甘草苷、异甘草素,该类化合物具有镇咳祛痰、抗炎、抗肿瘤、抗氧化、抗纤维化等多种药理活性。三萜皂苷类化合物为甘草酸,该化合物具有抗炎、解毒、免疫调节、抗病毒、保肝和稳定细胞膜等作用。上述化学成分均为甘草发挥其“清热解毒,祛痰止咳”功效和抗肿瘤、抗炎、抗菌、抗病毒、肝脏保护等药理作用的主要生物活性成分,说明本方法选择的特征峰能够较为科学地反映甘草药材的药效。
附图说明
18.图1是10批甘草供试药材的叠加图谱。
19.图2是对照药材特征图谱及供试药材共有模式。
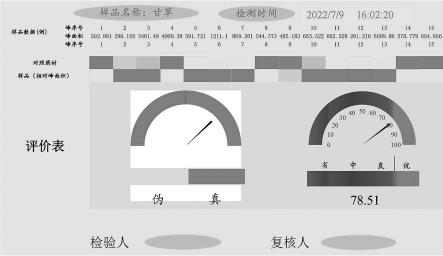
20.图3是新疆甘草样品质-量双标质量控制方法可视化结果。
具体实施方式
21.以下实施例将有助于对本发明的了解,但这些实施例仅为了对本发明加以说明,本发明并不限于这些内容。
22.实施例1。
23.甘草对照药材(批号:120904-201620,中国食品药品检定研究所)、甘草药材(安徽、甘肃、湖北、吉林、内蒙、宁夏、山西、陕西、新疆、云南)经辽宁中医药大学中药鉴定教研室许亮教授鉴定为豆科植物甘草glycyrrhiza uralensis fisch.的根及根茎。根据2020版《中华人民共和国药典》甘草“含量测定”项下方法检测,10批产地供试品均能符合药典规定(含甘草苷不得少于0.5 %,甘草酸不得少于2.0%)。
24.1.基于对照药材的甘草药材定性研究。
25.1.1溶液的制备。
26.取甘草对照药材粉末0.50 g,精密称定,置5 ml容量瓶中,70%乙醇稀释至刻度,称定重量,超声(功率200 w,频率40 khz)30 min,放冷,再称定重量,用70%乙醇补足失重,摇匀,过0.22 μm微孔滤膜过滤,取续滤液,作为参照物溶液;取供试药材粉末(过40目筛)0.50 g,按上述方法制成供试品溶液。
27.1.2色谱条件。
28.色谱柱:agilent sb-c
18
色谱柱(4.6
×
100 mm,2.7-micron);流动相:0.1%甲酸水(a)-乙腈(b),进行梯度洗脱;梯度洗脱程序:0~3 min,10%
→
20%b;3~7 min,20%
→
20%b;7~9 min,20%
→
26%b;9~13 min,26%
→
26%b;13~15 min,26%
→
34%b;15~19 min,34%
→
36%b;19~22 min,36%
→
44%b;22~28 min,44%
→
48%b;28~40 min,48%
→
70%b。流速:0.8 ml/min;柱温:30℃;dad检测器波长:237 nm;进样量:5 μl。
29.1.3 特征图谱的建立。
30.将参照物溶液和供试品溶液,每份样品平行2次,按色谱条件依次进样检测,将237 nm波长下的图谱数据由分析检测仪器中导出。10批供试药材的hplc叠加图谱,见图1;10批供试药材使用中位数进行自动匹配,加以多点校正,生成共有模式r,与对照药材特征图谱进行比对,见图2。
31.1.4 相似度评价。
32.供试药材与对照药材的相似度均大于0.96。
33.1.5 特征峰化学成分解析。
34.采用q-tof-ms技术,通过分析化合物的保留时间、一级离子质荷比以及二级离子碎片信息,并与相关文献报道信息进行匹配,共鉴定出6个特征峰。
35.本发明所述的甘草药材质量控制方法,得到的甘草药材特征图谱可以用于判断甘草药材的真伪。
36.2.基于内标物质的特征峰化学成分相对定量研究。
37.13号峰“甘草酸”分离效果好、峰面积大且保留时间稳定居中,故以其作为内标物质,开展基于内标物质的特征峰相对定量研究。根据2020版《中华人民共和国药典》甘草“含量测定”项下方法检测,10批产地供试品均符合药典规定。通过内标物质化学成分准确定量,计算10批供试药材特征峰化学成分的相对含量。取“中位数-标准差”作为特征峰化学成分相对于内标物质化学成分的相对含量下限,见表1。
38.表1 基于内标物质的特征峰相对定量结果。
39.3.方法学考察。
40.3.1 精密度试验。
41.精密吸取同一供试品溶液,按色谱条件测定,连续进样6次,测定各色谱峰相对保留时间、相对峰面积,计算相对标准偏差。各色谱峰相对保留时间、相对峰面积rsd值均小于1.0%,表明仪器精密度良好。
42.3.2 稳定性试验。
43.精密吸取同一供试品溶液,按色谱条件,分别在0、2、4、8、12、24 h进样6次检测,测定各色谱峰相对保留时间、相对峰面积,计算相对标准偏差。各色谱峰相对保留时间、相对峰面积rsd值均小于1.2%,表明供试品溶液在24 h内稳定。
44.3.3 重复性实验。
45.按供试品溶液制备方法制备6份供试样品,按色谱条件,分别进样检测,测定各色谱峰相对保留时间、相对峰面积,计算相对标准偏差。各色谱峰相对保留时间、相对峰面积rsd值均小于2.1 %,表明方法重复性良好。
46.3.4 线性关系考察。
47.配制“甘草酸”的6个质量浓度溶液,按色谱条件,分别进样检测,以质量(x)为横坐
标,峰面积(y)为纵坐标,绘制标准曲线并进行线性回归,得到线性回归方程(y=3161.8x 30.754)和相关系数(r=0.9996)。结果表明“甘草酸”在0.0550-0.3300 mg范围内线性关系良好。
48.实施例2新疆甘草供试品的鉴别。
49.新疆甘草样品与对照药材的相似度为0.981。通过本发明所设计的中药材(饮片)可视化的质-量双标质量控制方法程序(01版),可以直观的判定新疆甘草样品为“真”,质量为“优”,见图3。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。