1.本发明涉及一种检测头孢克肟溶出度的方法,属于药物制剂分析检测领域。
背景技术:
2.头孢克肟(cefixime)为第三代口服头孢菌素类抗生素,临床上用于治疗由于敏感菌所引起的呼吸道系统感染、尿道感染、胆道感染等,剂型包括胶囊剂、片剂、颗粒剂、干混悬剂等。头孢克肟几乎不溶于水,目前,其制剂质量标准中的溶出度试验仅为单一溶出介质、单点测定,方法具有一定的局限性,区分力较弱,并不能全面反映产品的内在质量,为保证产品质量和疗效与原研药品一致,产品在开发过程需要进行一致性评价研究,其中对多个介质的溶出曲线研究是一致性评价的关键评价指标之一,贯穿研发、生产控制、变更等多个阶段,因此,溶出度的分析测试任务较以往增加数百倍甚至上千倍。
3.目前药典收载的头孢克肟制剂质量标准中的溶出度检测方法采用紫外-可见分光光度法,该法需要稀释,操作繁琐,不适合高通量样品的检测,且准确度有限,同时制剂中的色素和有关物质对紫外-可见分光光度法测定溶出度存在较大干扰。另有药典收载的头孢克肟制剂质量标准中的溶出度检测方法与其含量测定方法一致,系采用高效液相法进行测定,色谱条件为以十八烷基硅烷键合硅胶为填充剂的色谱柱,水系流动相(1.92%四丁基氢氧化铵溶液,ph调至6.5),该法对硅胶色谱柱损耗较大,高通量进样下色谱柱使用寿命较短,且单样检测时间较长,约15~20分钟/样。由于头孢克肟供试品的溶液稳定性较差,上述的操作条件下,无法满足高通量溶出度样品溶液稳定性的要求,因而不适合高通量溶出样品的测定。
4.因此,提供一种检测头孢克肟溶出度的方法,能够实现检测时间短、效率高、对色谱柱损耗低成为了本领域技术人员亟待解决的问题。
技术实现要素:
5.本发明旨在至少解决现有技术中存在的技术问题之一。为此,本发明提出一种检测头孢克肟溶出度的方法,具有操作简单、准确度高、单样检测时间短、检测效率高、对色谱柱损耗低、有机试剂消耗量少的优点。
6.根据本发明的第一方面实施例,提出了一种检测头孢克肟溶出度的方法,采用液相色谱通过反相色谱法进行检测,检测条件包括:
7.流动相包括由a相和b相混合而得的混合溶液,a相包括卤代有机酸水溶液,b相包括乙腈和低级醇中的至少一种;
8.所述流动相中a相和b相的体积比为65~95:5~35;
9.色谱柱为核壳型c18柱。
10.根据本发明第一方面的实施例的方法,至少具有如下有益效果:
11.1、本方法的检测条件下,单针检测时间短,可极高效率分析高通量溶出样品;
12.2、本方法的流动相体系对色谱柱填料损耗较小,延长了色谱柱使用周期等,大大
降低检测成本。
13.3、本方法的检测条件下,专属性高,不受头孢克肟的制剂辅料的干扰,可以同时满足多种溶出介质的供试品溶液的分离度要求。
14.4、本发明选用核壳型c18色谱柱协同流动相作用下,达到了对于头孢克肟溶出度检测的快速、高分辨率的分离效果。
15.5、本方法的流动相配制简单,操作的重现性高。
16.在本发明的本方法中通过对流动相体系进行了筛选和优化,大大缩短了检测时间,同时能有效降低色谱柱的损耗,从而大大降低了检测成本。
17.根据本发明的一些实施例,所述流动相中a相和b相的体积比为75~86:14~25。
18.根据本发明的一些优选地实施例,所述流动相中a相和b相的体积比为78~84:16~22。
19.根据本发明的一些实施例,所述a相选自1~3级卤代有机酸的水溶液。
20.根据本发明的一些优选地实施例,所述卤代有机酸水溶液中的有机酸选自1~3碳的低级酸。
21.根据本发明的一些实施例,所述1~3级卤代有机酸的水溶液的质量浓度为0.01%~2%。
22.根据本发明的一些实施例,所述a相选自三级卤代有机酸的水溶液。
23.根据本发明的一些优选地实施例,所述a相选自三级卤代乙酸的水溶液。
24.根据本发明的一些优选地实施例,所述三级卤代乙酸的水溶液的质量浓度为0.1%~1.5%。
25.根据本发明的一些优选地实施例,所述a相选自三氟乙酸的水溶液。
26.根据本发明的一些优选地实施例,所述三氟乙酸的水溶液的质量浓度为0.5%~1.0%。
27.根据本发明的一些实施例,所述低级醇选自1~4碳的低级醇。
28.根据本发明的一些优选地实施例,所述1~4碳的低级醇选自甲醇。
29.根据本发明的一些优选地实施例,所述b相选自乙腈。
30.根据本发明的一些优选地实施例,所述核壳型c18柱选自thermo accucore c18,phenomenex kinetex c18和agilent poroshell 120ec-c18中的至少一种。
31.根据本发明的一些优选地实施例,所述thermo accucore c18的规格为50mm*4.6mm*2.6μm。
32.根据本发明的一些优选地实施例,所述phenomenex kinetex c18的规格为50mm*4.6mm*2.6μm。
33.根据本发明的一些优选地实施例,所述agilent poroshell 120ec-c18的规格为50mm*4.6mm*2.6μm。
34.根据本发明的一些实施例,所述检测条件还包括以下i-v中的至少一项:
35.i.色谱柱规格为填料粒径不大于3μm;
36.ii.柱温30~60℃;
37.iii.流速1.0~2.0ml/min;
38.iv.采用紫外检测器进行检测,检测波长为254nm。
39.根据本发明的一些优选地实施例,所述柱温为30~60℃。
40.根据本发明的一些优选地实施例,所述柱温为40~50℃。
41.根据本发明的一些优选地实施例,所述柱温在45~50℃。
42.根据本发明的一些优选地实施例,所述流速为1.0~2.0ml/min。
43.根据本发明的一些优选地实施例,所述流速为1.2~1.8ml/min。
44.根据本发明的一些实施例,所述检测的方法包括以下步骤:
45.s1、制备供试品溶液和对照品溶液;
46.s2、通过液相色谱对所述供试品溶液和对照品溶液进行检测;
47.s3、对检测结果进行分析。
48.根据本发明的一些实施例,步骤s1中,所述供试品溶液的制备包括以下步骤:
49.取头孢克肟制剂,采用浆法/篮法,转速50~150r/min,进行溶出试验,在不同时间点取溶出液作供试品溶液。
50.根据本发明的一些实施例,步骤s1中,所述对照品溶液的制备包括以下步骤:
51.取头孢克肟对照品适量,以溶出介质为溶剂,溶解并稀释制成每1ml中约含头孢克肟(按c
16h15
n5o7s2计)50~200μg的溶液,作为对照品溶液。
52.根据本发明的一些实施例,步骤s2中,所述对供试品溶液和对照品溶液通过液相色谱进行检测包括以下步骤:
53.取供试品溶液、对照品溶液各5μl,分别注入液相色谱仪,按照所述色谱条件进行测定,记录色谱图,按外标法以峰面积计算头孢克肟的溶出度。
[0054][0055][0056]
式中:
[0057]
f:对照品溶液中头孢克肟的校正因子;
[0058]
对照品溶液中头孢克肟的校正因子f的平均值;
[0059]
wstd:对照品溶液中头孢克肟对照品的称样量,mg;
[0060]
p:头孢克肟对照品的含量;
[0061]
astd:对照品溶液中头孢克肟的峰面积;
[0062]
dstd:对照品溶液体积,ml;
[0063]
aspl:供试品溶液中头孢克肟的峰面积;
[0064]
dspl:供试品溶液体积,ml。
[0065]
lc:头孢克肟制剂的标示量,mg。
[0066]
根据本发明的一些实施例,所述供试品选自头孢克肟颗粒、头孢克肟胶囊、头孢克肟片、头孢克肟分散片和头孢克肟干混悬剂中的至少一种。
[0067]
根据本发明的一些实施例,所述溶出介质包括ph1.2盐酸溶液。
[0068]
根据本发明的一些实施例,所述ph1.2盐酸溶液的配制方法包括:取氯化钠2g,加盐酸7ml,再加纯化水稀释至1000ml,即得。
[0069]
根据本发明的一些实施例,所述溶出介质包括ph4.5醋酸盐缓冲液。
[0070]
根据本发明的一些实施例,所述ph4.5醋酸盐缓冲液的配制方法包括:取醋酸钠18g,加冰醋酸9.8ml,再加纯化水稀释至1000ml,即得。
[0071]
根据本发明的一些实施例,所述溶出介质包括ph6.8磷酸盐缓冲液。
[0072]
根据本发明的一些实施例,所述ph6.8磷酸盐缓冲液的配制方法包括:取3.40磷酸二氢钾、3.55g无水磷酸氢二钠,用纯化水稀释至1000ml,摇匀,即得。
[0073]
根据本发明的一些实施例,所述溶出介质包括ph7.2磷酸盐缓冲液。
[0074]
根据本发明的一些实施例,所述ph7.2磷酸盐缓冲液的配制方法包括:取0.2mol/l磷酸二氢钾溶液50ml与0.2mol/l氢氧化钠溶液35ml,加新沸过的冷水稀释至200ml,摇匀,即得。
[0075]
根据本发明的一些实施例,所述溶出介质包括ph7.5磷酸盐缓冲液。
[0076]
根据本发明的一些实施例,所述ph7.5磷酸盐缓冲液的配制方法包括:取0.05mol/l磷酸氢二钠溶液1000ml、0.025mol/l枸橼酸溶液1000ml,混匀,用氢氧化钠调ph至7.5,即得。
[0077]
根据本发明的一些实施例,所述溶出介质包括纯化水。
[0078]
根据本发明的一些实施例,所述供试品溶液的制备包括:取头孢克肟颗粒,照溶出度法(中国药典2020版四部通则0931第二法),以ph1.2盐酸溶液900ml为溶出介质,转速为50r/min,依法操作,于溶出终点取溶出液适量,过滤,取续滤液作为供试品头孢克肟ph1.2盐酸溶液。同法制得以ph 4.5醋酸盐缓冲液、ph 6.8磷酸盐缓冲液、ph 7.2磷酸盐缓冲液、ph 7.5磷酸盐缓冲液、纯化水为溶出介质的供试品。纯化水、ph1.2盐酸溶液、ph 4.5醋酸盐缓冲液介质的溶出终点为120min,ph6.8磷酸盐缓冲液、ph7.2磷酸盐缓冲液、ph7.5磷酸盐缓冲液介质的溶出终点为30min。
[0079]
根据本发明的一些实施例,所述对照品溶液的制备包括:取头孢克肟对照品适量,加ph1.2的盐酸溶液溶解并稀释制成每1ml中约含头孢克肟(按c
16h15
n5o7s2计)50μg的溶液,作为对照品溶液。同法制得以ph 4.5醋酸盐缓冲液、ph6.8磷酸盐缓冲液、ph 7.2磷酸盐缓冲液、ph 7.5磷酸盐缓冲液、纯化水为溶出介质的对照品溶液。
[0080]
根据本发明的一些实施例,所述空白辅料溶液的制备包括:称取头孢克肟颗粒空白辅料95mg至100ml量瓶中,加ph1.2盐酸溶液溶解并定容,过滤,取续滤液作为空白辅料溶液。同法制得以ph4.5醋酸盐缓冲液、ph6.8磷酸盐缓冲液、ph7.2磷酸盐缓冲液、ph7.5磷酸盐缓冲液、纯化水为溶出介质的空白辅料溶液。
[0081]
上述空白辅料包括:蔗糖、日落黄、黄原胶、阿拉伯胶、瓜尔胶、西黄蓍胶、羟丙基纤维素和甜橙粉末香精。
[0082]
根据本发明的一些实施例,所述步骤s3中的分析包括定性和/或定量分析。
[0083]
根据本发明的一些实施例,所述分析采用的方法选自面积归一化法、自身对照法、内标法或外标法中的至少一种。
[0084]
本发明的其它特征和优点将在随后的说明书中阐述,并且,部分地从说明书中变得显而易见,或者通过实施本发明而了解。
附图说明
[0085]
本发明的上述和/或附加的方面和优点从结合下面附图对实施例的描述中将变得明显和容易理解,其中:
[0086]
图1为本发明实施例1溶出介质hplc色谱图;
[0087]
图2为本发明实施例1空白辅料溶液hplc色谱图;
[0088]
图3为本发明实施例1对照品溶液hplc色谱图;
[0089]
图4为本发明实施例1供试品溶液hplc色谱图;
[0090]
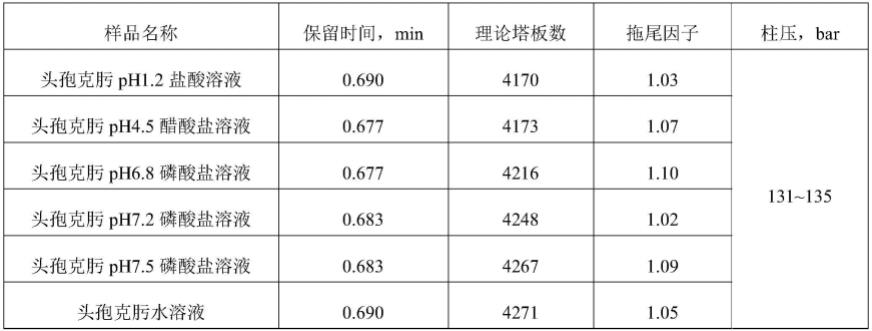
图5为本发明实施例1系统适用性hplc色谱图;
[0091]
图6为本发明实施例2供试品溶液及空白辅料溶液hplc色谱图;
[0092]
图7为本发明实施例3供试品溶液及空白辅料溶液hplc色谱图;
[0093]
图8为本发明实施例4供试品溶液及空白辅料溶液hplc色谱图;
[0094]
图9为本发明实施例5供试品溶液及空白辅料溶液hplc色谱图;
[0095]
图10为本发明实施例6供试品溶液及空白辅料溶液hplc色谱图
[0096]
图11为本发明实施例7供试品溶液及空白辅料溶液hplc色谱图;
[0097]
图12为本发明实施例8、实施例9系统适用性溶液hplc色谱图;
[0098]
图13为本发明测试例1标准曲线图;
[0099]
图14为本发明测试例1标准曲线图;
[0100]
图15为本发明测试例1标准曲线图;
[0101]
图16为本发明测试例1标准曲线图;
[0102]
图17为本发明测试例1标准曲线图;
[0103]
图18为本发明测试例1标准曲线图;
[0104]
图19为本发明对比例1系统适用性溶液hplc色谱图;
[0105]
图20为本发明对比例1系统适用性溶液hplc色谱图;
[0106]
图21为本发明对比例2供试品溶液hplc色谱图;
[0107]
图22为本发明对比例2供试品溶液hplc色谱图;
[0108]
图23为本发明对比例3不同检测方法的供试品溶液hplc色谱对比图。
具体实施方式
[0109]
下面详细描述本发明的实施例,所述实施例的示例在附图中示出,其中自始至终相同或类似的标号表示相同或类似的元件或具有相同或类似功能的元件。下面通过参考附图描述的实施例是示例性的,仅用于解释本发明,而不能理解为对本发明的限制。
[0110]
在本发明的描述中,如果有描述到第一、第二等只是用于区分技术特征为目的,而不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量或者隐含指明所指示的技术特征的先后关系。
[0111]
在本发明的描述中,需要理解的是,涉及到方位描述,例如上、下等指示的方位或位置关系为基于附图所示的方位或位置关系,仅是为了便于描述本发明和简化描述,而不是指示或暗示所指的装置或元件必须具有特定的方位、以特定的方位构造和操作,因此不能理解为对本发明的限制。
[0112]
本发明的描述中,需要说明的是,除非另有明确的限定,设置、安装、连接等词语应
做广义理解,所属技术领域技术人员可以结合技术方案的具体内容合理确定上述词语在本发明中的具体含义。
[0113]
实施例1
[0114]
本实施例提供了一种头孢克肟制剂溶出度的快速测定方法,具体为:
[0115]
溶出介质的配制:
[0116]
①
、ph1.2盐酸溶液溶出介质的制备:取氯化钠2g,加盐酸7ml,再加纯化水稀释至1000ml,即得。
[0117]
②
、ph4.5醋酸盐缓冲液溶出介质的制备:取醋酸钠18g,加冰醋酸9.8ml,再加纯化水稀释至1000ml,即得。
[0118]
③
、ph6.8磷酸盐缓冲液溶出介质的制备:取3.40磷酸二氢钾、3.55g无水磷酸氢二钠,用纯化水稀释至1000ml,摇匀,即得。
[0119]
④
、ph7.2磷酸盐缓冲液溶出介质的制备:取0.2mol/l磷酸二氢钾溶液50ml与0.2mol/l氢氧化钠溶液35ml,加新沸过的冷水稀释至200ml,摇匀,即得。
[0120]
⑤
、ph7.5磷酸盐缓冲液溶出介质的制备:取0.05mol/l磷酸氢二钠溶液1000ml、0.025mol/l枸橼酸溶液1000ml,混匀,用氢氧化钠调ph至7.5,即得。
[0121]
⑥
、纯化水(符合2020版中国药典规定)。
[0122]
供试品溶液的制备:取头孢克肟颗粒(50mg,批号:g1802001),照溶出度法(中国药典2020版四部通则0931第二法),以ph1.2盐酸溶液900ml为溶出介质,转速为50r/min,依法操作,于溶出终点取溶出液1.5ml,过滤,取续滤液作为头孢克肟ph1.2盐酸介质供试品溶液。同法制得以ph 4.5醋酸盐缓冲液、ph 6.8磷酸盐缓冲液、ph 7.2磷酸盐缓冲液、ph 7.5磷酸盐缓冲液、纯化水为溶出介质的头孢克肟供试品溶液。纯化水、ph1.2盐酸溶液、ph 4.5醋酸盐缓冲液介质的溶出终点为120min,ph6.8磷酸盐缓冲液、ph7.2磷酸盐缓冲液、ph7.5磷酸盐缓冲液介质的溶出终点为30min。
[0123]
对照品溶液的制备:精密称取头孢克肟对照品适量(按c
16h15
n5o7s2计约5~20mg),置于100ml的量瓶中,加ph1.2的盐酸溶液溶解并稀释制成每1ml中约含50μg~200μg头孢克肟的溶液(根据制剂标示量配制相应浓度的对照品溶液),作为ph1.2盐酸介质对照品溶液。同法制得以ph 4.5醋酸盐缓冲液、ph6.8磷酸盐缓冲液、ph 7.2磷酸盐缓冲液、ph 7.5磷酸盐缓冲液、纯化水为溶出介质的对照品溶液。
[0124]
空白溶剂的制备:以实际溶出介质作为空白溶剂。
[0125]
空白辅料溶液的制备:称取头孢克肟颗粒空白辅料95mg至100ml量瓶中,加ph1.2盐酸溶液溶解并定容,过滤,取续滤液作为ph1.2盐酸介质空白辅料溶液。同法制得以ph4.5醋酸盐缓冲液、ph6.8磷酸盐缓冲液、ph7.2磷酸盐缓冲液、ph7.5磷酸盐缓冲液、纯化水为溶出介质的空白辅料溶液。
[0126]
系统适用性溶液的制备:取头孢克肟对照品适量,加水溶解并稀释制成每1ml中约含头孢克肟1mg的溶液,于95℃水浴上加热45分钟,冷却至室温,作为系统适用性溶液。
[0127]
检测:通过液相色谱仪进行检测,条件如下:
[0128]
色谱柱:thermo accucore c18,50mm*4.6mm*2.6μm;流动相:以0.8%三氟乙酸水溶液-乙腈(80:20)为流动相;柱温45℃;流速1.2ml/min;检测波长254nm。
[0129]
取溶出介质、空白辅料溶液、对照品溶液、供试品溶液及系统适用性溶液各5μl注
入液相色谱仪,记录色谱图。所得色谱图如图1-5所示,供试品色谱数据如表1所示:
[0130]
表1六种溶出介质中头孢克肟溶出度供试品的高效液相色谱分析数据表
[0131][0132]
图1、图2表明六种空白溶剂和六种介质配制的空白辅料溶液在实施例方法下没有相关色谱峰,对主峰没有干扰;
[0133]
图3为实施例1对照品溶液hplc色谱图(从上到下依次为ph1.2盐酸溶液、ph4.5醋酸盐溶液、ph 6.8磷酸盐溶液、ph 7.2磷酸盐溶液、ph 7.5磷酸盐溶液、纯水配制的头孢克肟对照品溶液);
[0134]
图4为本发明实施例1供试品溶液hplc色谱图(从上到下依次为ph1.2盐酸溶液、ph4.5醋酸盐溶液、ph6.8磷酸盐溶液、ph7.2磷酸盐溶液、ph7.5磷酸盐溶液、纯水配制的头孢克肟供试品溶液);
[0135]
图3和图4表明使用上述介质配制的对照品溶液和供试品溶液中头孢克肟的保留时间一致,本方法专属性良好;
[0136]
图5为本发明实施例1系统适用性hplc色谱图;为采用药典中含量测定方法中规定的系统适用性溶液在本方法下进行的表征,结果表明,头孢克肟表明主峰与头孢克肟杂质d峰的分离度良好,其中,头孢克肟的出锋时间为0.69min,杂质d出锋的时间为0.817min。
[0137]
以上结果表明,采用本发明方法能同时满足多个溶出介质的供试品溶液的检测分离要求,专属性良好,单样检测时间小于1min,极大提升检测效率。
[0138]
实施例2
[0139]
本实施例提供了一种头孢克肟制剂溶出度的快速测定方法,具体步骤为:
[0140]
采用实施例1中的ph1.2盐酸介质供试品溶液和ph1.2盐酸介质空白辅料溶液。测定条件与实施例1相比,检测柱温为35℃,其余条件均相同。所得供试品及空白辅料溶液色谱图如附图6。
[0141]
表2不同柱温条件头孢克肟供溶出度供试品的高效液相色谱分析数据表
[0142]
柱温,℃保留时间,min理论板数拖尾因子350.69742821.01
[0143]
结果表明,本实施例中头孢克肟的保留时间较实施例1略有延迟,空白辅料溶液中无干扰峰,表明该柱温范围内可满足快速检测需求。
[0144]
实施例3
[0145]
本实施例提供了一种头孢克肟制剂溶出度的快速测定方法,具体步骤为:
[0146]
采用实施例1中的ph1.2盐酸介质供试品溶液和ph1.2盐酸介质空白辅料溶液。测
定条件与实施例1相比,检测柱温为55℃,其余条件均相同。所得供试品及空白辅料溶液色谱图如图7所示(柱温55℃,主峰rt:0.683min)。
[0147]
表3头孢克肟供溶出度供试品的高效液相色谱分析数据表
[0148]
柱温,℃保留时间,min理论板数拖尾因子550.68341601.08
[0149]
表3结果表明,本实施例中头孢克肟的保留时间较实施例1略有提前,空白辅料溶液中无干扰峰,表明该柱温范围内可满足快速检测需求。结合实施例2结果,柱温在该范围内波动对头孢克肟的保留时间没有明显影响,但在实际操作中,55℃的柱温远高于室温,高温增加设备能耗,同时也对色谱柱内填料间的氢键造成影响、缩短色谱柱的使用周期。
[0150]
实施例4
[0151]
本实施例提供了一种头孢克肟制剂溶出度的快速测定方法,具体步骤为:
[0152]
采用实施例1中的ph1.2盐酸介质供试品溶液和ph1.2盐酸介质空白辅料溶液。测定条件与实施例1相比,流动相比例不同为0.8%三氟乙酸水溶液-乙腈(65:35),其余条件均相同。所得供试品及空白辅料溶液色谱图如图8所示(35乙腈%,主峰rt:0.573min)。
[0153]
表4不同比例流动相头孢克肟供溶出度供试品的高效液相色谱分析数据表
[0154]
流动相比例保留时间,min柱压,bar理论板数拖尾因子0.8%三氟乙酸水溶液-乙腈(65:35)0.57320038971.03
[0155]
实施例5
[0156]
本实施例提供了一种头孢克肟制剂溶出度的快速测定方法,具体步骤为:
[0157]
采用实施例1中的ph1.2盐酸介质供试品溶液和ph1.2盐酸介质空白辅料溶液。测定条件与实施例1相比,流动相比例不同为0.8%三氟乙酸水溶液-乙腈(95:5),其余条件均相同。所得供试品及空白辅料溶液色谱图如图9所示(乙腈5%,主峰rt:0.897min)。
[0158]
表5不同比例流动相头孢克肟供溶出度供试品的高效液相色谱分析数据表
[0159]
流动相比例保留时间,min柱压,bar理论板数拖尾因子0.8%三氟乙酸水溶液-乙腈(95:5)0.89715044031.06
[0160]
表4,5结果表明,本实施例中头孢克肟的保留时间受有机相比例影响较大,乙腈比例越高,主峰洗脱出峰时间越早;空白辅料溶液中无干扰峰,表明该流动相比例范围内可满足快速检测需求。但在实际操作中,实施例4方法的流动相比例在所有比例范围内记录到最高柱压,长时间的高压力容易冲塌色谱柱柱头填料、造成损伤,对比实施例1使用更多的有机相;
[0161]
实施例5方法中乙腈比例为5%时,头孢克肟保留时间较实施例1延迟30%,实施例1的检测效率更优。
[0162]
实施例6
[0163]
本实施例提供了一种头孢克肟制剂溶出度的快速测定方法,具体步骤为:
[0164]
采用实施例1中的ph1.2盐酸介质供试品溶液和ph1.2盐酸介质空白辅料溶液。测定条件与实施例1相比,流速不同为1.0ml/min,其余条件均相同。所得色谱图如图10(流速1.0ml/min,主峰rt:0.737min)所示。
[0165]
表6不同流速下头孢克肟溶出液的高效液相色谱分析数据表
[0166]
流速,ml/min保留时间,min理论塔板数拖尾因子柱压1.00.73740441.10110
[0167]
表6结果表明,本实施例中头孢克肟的保留时间略有不同,空白辅料溶液中无干扰峰,表明该流速范围内可满足快速检测需求。
[0168]
实施例7
[0169]
本实施例提供了一种头孢克肟制剂溶出度的快速测定方法,具体步骤为:
[0170]
采用实施例1中的ph1.2盐酸介质供试品溶液和ph1.2盐酸介质空白辅料溶液。测定条件与实施例1相比,流速不同为2.0ml/min,其余条件均相同。所得色谱图如图11(流速2.0ml/min,主峰rt:0.653min)所示。
[0171]
表7不同流速下头孢克肟溶出液的高效液相色谱分析数据表
[0172]
流速,ml/min保留时间,min理论塔板数拖尾因子柱压2.00.65343171.03210
[0173]
表7结果表明,本实施例中头孢克肟的保留时间略有不同,空白辅料溶液中无干扰峰,表明该流速范围内可满足快速检测需求。
[0174]
表6,7结果表明,本实施例中头孢克肟的保留时间受流速影响较大,流速越高,主峰洗脱出峰时间越早;空白辅料溶液中无干扰峰,表明该流速范围内可满足快速检测需求。但在实际操作中,实施例6方法中流速为1.0ml/min时,头孢克肟保留时间较实施例1延迟7%,实施例1的检测效率、理论塔板数和拖尾因子更优;实施例7方法中流速为2.0ml/min时,在所有流速内记录到最高柱压,在高通量检测环境下需要消耗更多的流动相。
[0175]
实施例8
[0176]
本实施例提供了一种头孢克肟制剂溶出度的快速测定方法,具体步骤为:
[0177]
采用实施例1中的系统适用性溶液,测定条件与实施例1相比,色谱柱为phenomenex kinetex c18核壳柱,其余条件均相同。所得色谱图如图12所示,测试结果如表8所示。
[0178]
实施例9
[0179]
本实施例提供了一种头孢克肟制剂溶出度的快速测定方法,具体步骤为:
[0180]
采用实施例1中的系统适用性溶液,测定条件与实施例1相比,色谱柱为agilent poroshell 120ec-c18核壳柱,其余条件均相同。所得色谱图如图12所示,测试结果如表8所示。
[0181]
实施例1中的系统适用性溶液,色谱柱为thermo accucore c18核壳柱,如图12所示。
[0182]
表8不同厂家色谱柱条件下系统适用性溶液测定结果
[0183]
[0184]
表8结果表明,本实施例中头孢克肟的保留时间有所不同,但均在1.5min内洗脱出峰,不同品牌的核壳型色谱柱均能满足快速检测需求。在同样的色谱条件下,agilent poroshell 120ec-c18色谱柱对头孢克肟和杂质d的分离效果最佳,实施例1选用色谱柱中头孢克肟保留时间最短,综合主峰理论塔板数、拖尾因子等因素考虑,优选实施例1中色谱柱。图12中,自上至下依次为thermo accucore c18色谱柱,phenomenex kinetex c18色谱柱和agilent poroshell 120ec-c18色谱柱的测试结果。
[0185]
测试例1
[0186]
为了考察本发明分析方法的准确性,对实施例1分析方法进行了方法学考察。
[0187]
一、线性相关性实验
[0188]
精密称取头孢克肟对照品适量(按c
16h15
n5o7s2计约100mg),置100ml量瓶中,加适量甲醇溶解,加水释至刻度,摇匀,作为线性试验对照品贮备液。
[0189]
精密量取对照品贮备液0.1、0.5、1.0、1.5、2.0、2.5ml,分别置10ml量瓶中,加入不同溶出介质稀释至刻度,摇匀,得系列浓度的线性溶液,过滤后分别取续滤液5μl注入液相色谱仪,记录色谱图。以头孢克肟的浓度c(μg/ml)为横坐标、峰面积(a)为纵坐标,绘制标准曲线。求得线性回归方程结果见表9,表明头孢克肟在10.05μg/ml~251.30μg/ml范围内,峰面积与浓度呈良好的线性关系。标准曲线图见图13~图18。
[0190]
表9头孢克肟在不同介质的线性回归结果
[0191][0192]
二、准确度实验
[0193]
分别向含有空白辅料的量瓶中加入一定量的头孢克肟对照品配制成50%、75%、100%浓度的准确度溶液,按照本发明检测方法进行分析,测得头孢克肟在不同介质的回收率,准确度实验结果见表10。
[0194]
表10头孢克肟在不同溶出介质的回收率结果
[0195]
[0196]
[0197][0198]
三、精密度实验
[0199]
取线性项下对照品贮备液0.5ml,分别置10ml量瓶中,加不同溶出介质稀释至刻度,摇匀,过滤后取续滤液5μl注入液相色谱仪,连续进样6次,计算头孢克肟峰面积rsd,结果见表11,测试方法精密度良好。
[0200]
表11进样精密度试验结果
[0201][0202]
四、重复性实验
[0203]
取实施例1中不同溶出介质的同一溶出杯中溶出液6份,过滤后取续滤液分别进样,计算溶出度,结果见表12,重复性良好。
[0204]
表12重复性试验结果
[0205][0206]
五、溶液稳定性实验
[0207]
取头孢克肟原料药粉末适量,分别用六种溶出介质配制成约含头孢克肟50ug/ml的溶液,分别放置于10℃、25℃,于0,2,4,6,8小时取5μl注入液相色谱仪,记录色谱图。以各时间点主峰峰面积计算rsd%,考察头孢克肟溶解于各溶出介质中的稳定性。结果见表13、表14,结果显示供试品溶液在各介质中,25℃条件与10℃条件下放置8h,峰面积的rsd均小
于1%,供试品溶液在各溶出介质中均较为稳定。
[0208]
表13头孢克肟在各溶出介质稳定性试验结果(10℃)
[0209][0210]
表14头孢克肟在各溶出介质稳定性试验结果(25℃)
[0211][0212]
六、色谱柱损耗实验
[0213]
启用新色谱柱(thermo accucore c18,50mm*4.6mm,2.6μm),取实施例1下ph1.2盐酸介质供试品溶液多次进样,考察色谱柱的损耗情况,结果见表15。结果显示,经约1000针进样后,主峰的拖尾因子和理论塔板数与首次进样相比,无明显变化;经约3000针进样后,主峰的理论塔板数开始锐减,通过测试表明,在本发明提供的色谱条件下,该核壳型色谱柱可供3000针以上的样品测试,适合高通量样品的检测。
[0214]
表15头孢克肟供试品溶液多次进样测定结果
[0215]
柱效参数首次进样进样约1000针进样约3000针拖尾因子1.071.051.22理论塔板数439641772825
[0216]
对比例1
[0217]
本对比例提供了一种头孢克肟制剂溶出度的测定方法,与实施例1相比,本对比例按照日本橙皮书收载的溶出度测定方法对系统适用性溶液进行测定,色谱条件为:以十八烷基硅烷键合硅胶为填充剂的色谱柱,水系流动相(1.92%四丁基氢氧化铵溶液,ph调至6.5):有机相(乙腈)比例为3:1,检测波长254nm,柱温40℃,流速1.5ml/min,测试结果如表
16所示:
[0218]
对比例1所得色谱图见图19(初次进样)和图20(进样约850针),经多次进样后,色谱图中头孢克肟峰理论塔板数显著降低,色谱峰出现拖尾、峰顶变圆等柱效下降的现象。系统适用性溶液中头孢克肟与杂质d的分离度也因为柱效的下降而不符合要求(二者分离度应≥1.5),表明本实施例方法不适合高通量样品的检测。其中,初次进样时,杂质的出锋时间为10.523min,头孢克肟的出锋时间为12.287min,二者分离度为3.67,头孢克肟峰理论塔板数为8457;进样850针的杂质的出锋时间为10.29min,头孢克肟的出锋时间为12.05min,二者分离度下降至1.29,头孢克肟峰理论塔板数下降至2893,色谱柱柱效与初次进样时相比显著下降。
[0219]
表16多次进样后系统适用性溶液测定结果
[0220]
头孢克肟峰初次进样进样约850针保留时间,min12.28712.05理论塔板数84572893拖尾因子1.331.87与杂质d分离度3.671.29
[0221]
对比例2
[0222]
本对比例提供了一种头孢克肟制剂溶出度的测定方法,对比例1相比,本对比例采用核壳型c18色谱柱,其余条件均相同,对ph1.2盐酸溶液溶出介质的供试品溶液进行测定,多次进样后的结果见表17,色谱图见图21(初次进样)和图22(进样300针)。经多次进样后,色谱图中头孢克肟峰理论塔板数显著降低,色谱峰出现拖尾、峰顶变圆等柱效下降的现象,不符合检测要求,表明本对比例的方法不适合高通量样品的检测。其中初次进样的头孢克肟的出锋时间为1.907min,理论塔板数为6448,主峰峰形正常,进样300针的头孢克肟的出锋时间为1.862min,理论塔板数为2997,主峰峰形异常,色谱柱柱效与初次进样时相比显著下降。
[0223]
表17多次进样后供试品溶液测定结果
[0224]
头孢克肟峰初次进样进样300针保留时间,min1.9071.862理论塔板数64482997拖尾因子1.190.716
[0225]
对比例3
[0226]
本对比例提供了一种头孢克肟制剂溶出度的测定方法的对比结果:
[0227]
采用实施例1中的ph1.2盐酸介质供试品溶液,与实施例1相比,本对比例的色谱柱为普通型c18色谱柱,其余条件均相同。对比结果如表18所示,色谱图见图23,自上至下依次为对比例4色谱条件(普通型c18色谱柱)得到的色谱图和对比例3色谱条件(普通型c18色谱柱)测试得到的色谱图。
[0228]
表18使用普通c18色谱柱的供试品溶液测定结果
[0229][0230]
对比例4
[0231]
本对比例提供了一种头孢克肟制剂溶出度的测定方法的对比结果:
[0232]
采用实施例1中的ph1.2盐酸介质供试品溶液,与对比例3相比,本对比例采用对比例1下的流动相条件进行试验,其余条件均相同。对比结果如表19所示,色谱图见图23,自上至下依次为对比例4色谱条件(普通型c18色谱柱)得到的色谱图和对比例3色谱条件(普通型c18色谱柱)测试得到的色谱图。
[0233]
表19使用普通c18色谱柱、不同方法的供试品溶液测定结果
[0234][0235]
按照对比例3的方法和对比例4色谱条件分别在同一普通型c18色谱柱中测定,与对比例4色谱条件相比,对比例3的色谱条件的头孢克肟保留时间提前60%,单样检测时间缩短至5~7min,但与实施例1(核壳型c18色谱柱)的头孢克肟保留时间(约0.690min)相比,单样检测耗时为实施例1的10倍以上,检测效率较低,不适合高通量样品的检测。
[0236]
本发明通过对检测条件的筛选和优化配合核壳型c18色谱柱,大大缩短了检测时间,同时能有效降低色谱柱的损耗,从而降低了检测成本。
[0237]
以上所述仅为本发明的实施例,并非因此限制本发明的专利范围,凡是利用本发明说明书及附图内容所作的等同变换,或直接或间接运用在相关的技术领域,均同理包括在本发明的专利保护范围内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。