1.本发明属于光催化技术领域,尤其涉及一种无定形两相异质结光催化剂及其原位合成方法。
背景技术:
2.目前,挥发性有机化合物(vocs),主要来自工业生产、运输、和室内硬件,对大气和人类健康构成重大威胁。甲苯,主要来自汽车尾气、汽油加工排放、溶剂损失和工业活动排放物,在空气中难以自然分解。vocs治理技术主要包括活性炭吸附、热催化氧化、生物处理、变压吸附分离与净化技术、冷凝回收等方法。热催化氧化技术具有启动时间快、净化效率高、技术成熟等优势,但热催化氧化技术运行成本高和额外的能量输入。相比热催化技术,光催化氧化技术反应条件温和,不需要额外的能量输入就可将污染物氧化和矿化,被认为是最具发展潜力的vocs治理技术。
3.光催化剂受光照激发产生光生电子-空穴对,迁移到催化剂表面的光生电子与氧气发生反应产生超氧自由基,光生空穴与水分子反应形成羟基自由基。具有强氧化能力的超氧自由基和羟基自由基进攻vocs分子使其矿化。羟基自由基(
·
oh)相较超氧自由基具有更强的氧化能力,对vocs降解和矿化尤为重要。羟基可来源于反应中的水分子和催化剂表面的羟基。除调控反应体系中的湿度,活化催化剂表面羟基可达到促进羟基自由基的产生效果,实现提升光催化剂vocs矿化的目的。
4.znsn(oh)6光催化剂表面具有丰富的羟基,且价带较正,有利于超氧自由基的产生。但znsn(oh)6光催化剂带隙宽,光生载流子分离效率低,导致了光催化活性低。在此,我们提出以痕量非晶态sno2表面修饰znsn(oh)6光催化剂的策略提升其光催化vocs降解性能。传统的异质结光催化剂需要两相分开合成,反应复杂,有严苛的晶格匹配要求。该策略可在合成znsn(oh)6光催化剂的同时原位在其表面生成痕量非晶态sno2,形成非晶态sno
2-znsn(oh)6光催化剂异质结。该催化剂制备方法和原位形成非晶态sno
2-znsn(oh)6光催化剂能够有效提高宽带隙金属氢氧化物活性和稳定性,同时保持宽带隙金属氢氧化物规则的形貌和具有丰富的表面羟基新型材料,不仅可以改变宽带隙金属氢氧化物在实际应用中载流子分离效率低的格局,还在方法学研究上具有理论意义,使得宽带隙金属氢氧化物材料具有广泛的应用前景。
5.通过上述分析,现有技术存在的问题及缺陷为:传统的异质结光催化剂需要两相分开合成,反应复杂,有严苛的晶格匹配要求;znsn(oh)6光催化剂存在带隙宽,光生载流子分离效率低,光催化活性低;传统的光催化剂的羟基来源于环境湿度,同时催化性能对反应湿度有较强的敏感性,较高或较低的工作湿度条件可导致催化剂的失活;贵金属负载可提高光催化剂的氧化能力,但是贵金属成本昂贵,在较高温度下极易出现催化剂烧结现象,不利于广泛的市场应用。
技术实现要素:
6.针对现有技术存在的问题,本发明提供了一种无定形两相异质结光催化剂及其原位合成方法,尤其涉及一种用于vocs降解的无定形原位生成宽带隙金属氢氧化物/氢氧化物sno2/znsn(oh)6的异质结光催化剂及其原位合成方法。
7.本发明是这样实现的,一种无定形两相异质结光催化剂的原位合成方法,所述无定形两相异质结光催化剂的原位合成方法包括:
8.将含zn可溶性盐和含sn可溶性盐分别溶于去离子水中,并将含sn可溶性盐溶液滴入含zn可溶性盐溶液,搅拌;置于聚四氟乙烯水热釜进行反应,并将反应产物洗涤,离心,干燥,制备得到无定形两相异质结光催化剂。
9.进一步,所述无定形两相异质结光催化剂的原位合成方法包括以下步骤:
10.步骤一,将含zn可溶性盐溶于去离子水中,剧烈搅拌使得zn可溶性盐充分完全溶解形成溶液a,浓度为umg/ml;
11.步骤二,将含sn可溶性盐溶于去离子水中,剧烈搅拌使得sn可溶性盐充分完全溶解形成溶液b,浓度为vmg/ml;
12.步骤三,将wml体积的溶液b逐滴滴入溶液a中,同时进行剧烈搅拌,并将溶液的ph值保持在x,形成溶液c;
13.步骤四,将混合后的反应溶液c倒入聚四氟乙烯水热釜中,盖紧盖子,放入y℃烘箱中,保温zh,反应结束后待降至室温,结束合成过程;
14.步骤五,将反应完成后的溶液用去离子水和乙醇多次洗涤离心,将最后一次洗涤后的沉淀干燥后,得原位生成无定形sno2/znsn(oh)6异质结光催化剂。
15.进一步,所述步骤一中的含zn可溶性盐为碳酸锌、氯化锌、硝酸锌或乙酸锌中的一种,所述含sn可溶性盐为锡酸钠、锡酸钾或四氯化锡中的一种。
16.进一步,所述步骤一中的含zn可溶性盐溶液a的浓度u=60.0mg/ml。
17.进一步,所述步骤二中的含sn可溶性盐溶液b的浓度v=22.7mg/ml。
18.进一步,所述步骤三中的将wml的溶液b逐滴滴入溶液a中,w为18、20、22、24或26ml;将溶液的ph值保持在x,x=10。
19.进一步,所述步骤四中的将混合后的反应溶液c倒入聚四氟乙烯水热釜中,放入y℃烘箱中,y为100、120或140;保温zh,z为12、24、36或48。
20.进一步,所述步骤五中的将最后一次洗涤后的沉淀放入60℃的烘箱中干燥10h后,得到原位生成无定形sno2/znsn(oh)6异质结光催化剂。
21.本发明的另一目的在于提供一种实施所述的无定形两相异质结光催化剂的原位合成方法制备得到的无定形两相异质结光催化剂,所述无定形两相异质结光催化剂由无定形sno2和znsn(oh)6两相构成异质结形式存在。
22.所述无定形两相异质结光催化剂以非晶态的形式存在,且在合成znsn(oh)6催化剂的同时形成无定形sno2/znsn(oh)6异质结。
23.本发明的另一目的在于提供一种所述的无定形两相异质结光催化剂在光催化降解苯系物中的应用。
24.结合上述的技术方案和解决的技术问题,请从以下几方面分析本发明所要保护的技术方案所具备的优点及积极效果为:
25.第一、针对上述现有技术存在的技术问题以及解决该问题的难度,紧密结合本发明的所要保护的技术方案以及研发过程中结果和数据等,详细、深刻地分析本发明技术方案如何解决的技术问题,解决问题之后带来的一些具备创造性的技术效果。具体描述如下:
26.为了克服宽带隙金属氢氧化物光生载流子分离效率低的问题和充分利用氢氧化物表面丰富的羟基,本发明提出了一种以痕量金属氧化物替代贵金属催化剂提升光催化活性,改变宽带隙金属氢氧化物的光学性质,抵抗环境湿度干扰,合成操作简便,反应条件温和的两相宽带隙金属氧化物/氢氧化物异质结光催化剂的制备方法。
27.本发明通过合成宽带隙金属氧化物/氢氧化物异质结光催化剂两相异质结,以无定形sno2/znsn(oh)6为具体实施例,发明了一种简便、有效的方法增强宽带隙金属氢氧化物光催化材料的光生载流子分离效率以及光催化vocs降解、矿化的活性。
28.该光催化剂由无定形sno2和znsn(oh)6两相构成异质结形式存在。该光催化剂中的sno2含量极低,以非晶态的形式存在,不会对znsn(oh)6结构和形态造成影响。进而保护和利用表面丰富的羟基。该合成制备方法可在合成znsn(oh)6催化剂的同时形成无定形sno2/znsn(oh)6异质结,不需引入其他元素,反应简单。
29.本发明提供的无定形sno2/znsn(oh)6异质结光催化剂能够应用于光催化降解苯系物,以廉价sno2替代贵金属催化剂,活性大大超过贵金属负载的催化剂,是纯相znsn(oh)6光催化剂的14倍。该异质结的成功构建,既保留并利用了纯相znsn(oh)6光催化剂表面丰富的羟基,又提高了电子-空穴的分离效率,增强了znsn(oh)6光催化剂材料的活性和稳定性。相对于其他专利所报道的异质结而言,本发明提供的原位生成sno2/znsn(oh)6异质结光催化剂,未引入新的元素,由一步反应形成,合成简单,成本低廉,反应条件温和。本发明使得宽带隙双金属氢氧化物znsn(oh)6材料具有广泛的应用前景,同时在方法学研究上具有理论意义。
30.第二,把技术方案看做一个整体或者从产品的角度,本发明所要保护的技术方案具备的技术效果和优点,具体描述如下:
31.为了解决znsn(oh)6光催化剂带隙宽,光生载流子分离效率低,光催化活性低的问题,本发明合成了一种新型的原位生成无定形sno2/znsn(oh)6宽带隙金属氧化物异质结,该异质结保留了金属氢氧化物规则的形貌和丰富的表面羟基,同时引入无定形sno2相,扩宽了光响应范围,提高了电子-空虚的转移性能和效率,增强了光催化vocs降解和矿化的活性。
32.传统的异质结光催化剂需要两相分开合成,反应复杂,由严苛的晶格匹配要求,相比而言,本发明提供的原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结光催化剂未引进新的物质,通过调控合成条件一步完成,合成简单,反应温和,成本低廉,具有制备方法的指导意义。
33.第三,作为本发明的权利要求的创造性辅助证据,还体现在以下几个重要方面:
34.本发明的技术方案转化后的预期收益和商业价值为:
35.本发明制备的光催化剂催化剂表面丰富的羟基有利于具有强氧化能力的羟基自由基的形成,较传统商业p25光催化剂具有更高的voc降解性能和稳定性。合成的光催化剂对抵抗工作环境湿度的变化具有良好的表现,可适应复杂的环境湿度条件,应用范围广。以廉价sno2替代传统贵金属催化剂(au,pd)用于vocs光催化降解,催化活性是贵金属负载催
化剂的3.5倍,是纯相znsn(oh)6光催化剂的14倍,降低光催化剂的制备成本和系统启动成本。
附图说明
36.为了更清楚地说明本发明实施例的技术方案,下面将对本发明实施例中所需要使用的附图做简单的介绍,显而易见地,下面所描述的附图仅仅是本发明的一些实施例,对于本领域普通技术人员来讲,在不付出创造性劳动的前提下还可以根据这些附图获得其他的附图。
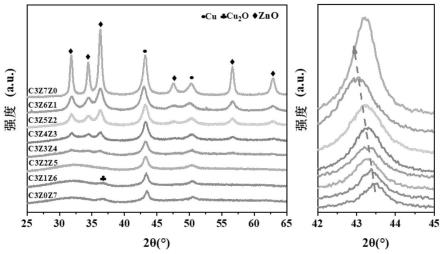
37.图1是本发明实施例1合成的纯相znsn(oh)6宽带隙金属氢氧化物光催化剂和实施例2在添加溶液b体积为20ml、22ml、24ml、26ml,水热温度为120℃,时间为24h,所合成的原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结光催化剂的xrd图;
38.图2是本发明实施例1合成的纯相znsn(oh)6宽带隙金属氢氧化物光催化剂和实施例2添加溶液b体积为24ml,水热温度为120℃,时间为24h,所合成的原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结和贵金属au(金)和pd(钯)负载的znsn(oh)6光催化剂的xrd图;
39.图3是本发明实施例1合成的znsn(oh)6宽带隙金属氢氧化物光催化剂的hrtem图;
40.图4是本发明实施例2添加溶液b体积为24ml,水热温度为120℃,时间为24h,所合成的原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结的tem图和hrtem图;
41.图5是本发明实施例1合成的纯相znsn(oh)6宽带隙金属氢氧化物光催化剂和实施例2在添加溶液b体积为20ml、22ml、24ml、26ml,水热温度为120℃,时间为24h,所合成的原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结光催化剂的光催化vocs降解和矿化的活性对比图;
42.图6本发明实施例2添加溶液b体积为24ml,水热温度为120℃,时间为24h,所合成的原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结在不同湿度条件下的光催化vocs降解和矿化的活性对比图;
43.图7是本发明实施例1合成的纯相znsn(oh)6宽带隙金属氢氧化物光催化剂和实施例2添加溶液b体积为24ml,水热温度为120℃,时间为24h,所合成的原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结和贵金属au(金)和pd(钯)负载的znsn(oh)6光催化剂的光催化vocs降解和矿化的活性对比图;
44.图8是本发明实施例1合成的纯相znsn(oh)6宽带隙金属氢氧化物光催化剂和实施例2在添加溶液b体积为24ml,水热温度为120℃,时间为24h,所合成的原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结光催化剂的xps图;
45.图9是本发明实施例1合成的纯相znsn(oh)6宽带隙金属氢氧化物光催化剂和实施例2在添加溶液b体积为24ml,水热温度为120℃,时间为24h,所合成的原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结光催化剂的uv-vis drs图;
46.图10是本发明实施例1合成的纯相znsn(oh)6宽带隙金属氢氧化物光催化剂和实施例2在添加溶液b体积为24ml,水热温度为120℃,时间为24h,所合成的原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结光催化剂的pl图;
47.图11是本发明实施例1合成的纯相znsn(oh)6宽带隙金属氢氧化物光催化剂和实
施例2在添加溶液b体积为24ml,水热温度为120℃,时间为24h,所合成的原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结光催化剂的光电流强度图;
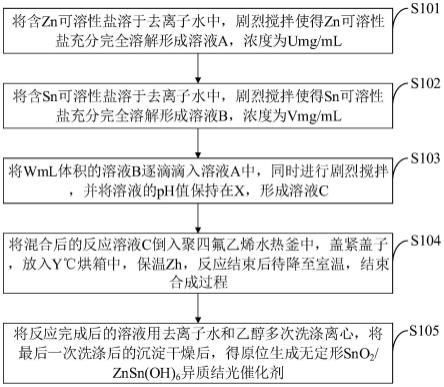
48.图12是本发明实施例提供的无定形两相异质结光催化剂及其原位合成方法流程图。
具体实施方式
49.为了使本发明的目的、技术方案及优点更加清楚明白,以下结合实施例,对本发明进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本发明,并不用于限定本发明。
50.针对现有技术存在的问题,本发明提供了一种无定形两相异质结光催化剂及其原位合成方法,下面结合附图对本发明作详细的描述。
51.一、解释说明实施例。为了使本领域技术人员充分了解本发明如何具体实现,该部分是对权利要求技术方案进行展开说明的解释说明实施例。
52.本发明实施例提供的无定形两相异质结光催化剂由无定形sno2和znsn(oh)6两相构成异质结形式存在。
53.本发明实施例提供的无定形两相异质结光催化剂以非晶态的形式存在,且在合成znsn(oh)6催化剂的同时形成无定形sno2/znsn(oh)6异质结。
54.如图12所示,本发明实施例提供的无定形两相异质结光催化剂的原位合成方法包括以下步骤:
55.s101,将含zn可溶性盐溶于去离子水中,剧烈搅拌使得zn可溶性盐充分完全溶解形成溶液a,浓度为umg/ml;
56.s102,将含sn可溶性盐溶于去离子水中,剧烈搅拌使得sn可溶性盐充分完全溶解形成溶液b,浓度为vmg/ml;
57.s103,将wml体积的溶液b逐滴滴入溶液a中,同时进行剧烈搅拌,并将溶液的ph值保持在x,形成溶液c;
58.s104,将混合后的反应溶液c倒入聚四氟乙烯水热釜中,盖紧盖子,放入y℃烘箱中,保温zh,反应结束后待降至室温,结束合成过程;
59.s105,将反应完成后的溶液用去离子水和乙醇多次洗涤离心,将最后一次洗涤后的沉淀干燥后,得原位生成无定形sno2/znsn(oh)6异质结光催化剂。
60.本发明实施例提供的步骤s101中的含zn可溶性盐为碳酸锌、氯化锌、硝酸锌或乙酸锌中的一种,所述含sn可溶性盐为锡酸钠、锡酸钾或四氯化锡中的一种;含zn可溶性盐溶液a的浓度u=60.0mg/ml。
61.本发明实施例提供的步骤s102中的含sn可溶性盐溶液b的浓度v=22.7mg/ml。
62.本发明实施例提供的步骤s103中的将wml的溶液b逐滴滴入溶液a中,w为18、20、22、24或26ml;将溶液的ph值保持在x,x=10。
63.本发明实施例提供的步骤s104中的将混合后的反应溶液c倒入聚四氟乙烯水热釜中,放入y℃烘箱中,y为100、120或140;保温zh,z为12、24、36或48。
64.本发明实施例提供的步骤s105中的将最后一次洗涤后的沉淀放入60℃的烘箱中干燥10h后,得到原位生成无定形sno2/znsn(oh)6异质结光催化剂。
65.二、应用实施例。为了证明本发明的技术方案的创造性和技术价值,该部分是对权利要求技术方案进行具体产品上或相关技术上的应用实施例。
66.所图5所示,是本发明实施例1合成纯相znsn(oh)6宽带隙金属氢氧化物光催化剂和实施例2在添加溶液b体积为20ml、22ml、24ml和26ml,水热温度为120℃,时间为24h,所合成的原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结光催化剂的光催化vocs降解和矿化的活性对比图。随着sno2量的增加,异质结光催化剂的光催化vocs降解率逐渐增加。性能最优样品为zsh24,其光催化vocs降解率为72%,是纯相的znsn(oh)6催化活性的15倍。将性能最优的sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结在不同湿度条件下的光催化vocs降解和矿化的活性对比图;相较相对湿度50%的反应条件,干燥空气测试条件下的sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结的光催化vocs降解率并未发生改变,表明sno2/znsn(oh)6异质结的表面羟基提供了反应所需羟基,同时具有良好的抵抗湿度变化的特性。
67.为与贵金属(金au,钯pd)负载的光催化剂作比较,将表面负载了贵金属au和pd的znsn(oh)6与sno2/znsn(oh)6异质结进行活性对比测试。如图7所示,原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结和贵金属au(金)和pd(钯)负载的znsn(oh)6光催化剂的光催化vocs降解和矿化的活性对比图。au/znsn(oh)6光催化剂的vocs降解效率为20.1%,而pd/znsn(oh)6光催化剂未表现出光催化vocs降解性能。结果表明,sno2/znsn(oh)6异质结光催化vocs降解性能分别是au/znsn(oh)6光催化剂的3.5倍,较贵金属光催化剂具有更优的光催化vocs降解活性。进一步说明,表面原位生成的sno2/znsn(oh)6合成操作简便,反应条件温和,具有有较强的光催化vocs降解能力、良好的抵抗工作湿度变化能力、较强的光催化稳定性以及可替代贵金属催化剂的低廉成本优势。
68.三、实施例相关效果的证据。本发明实施例在研发或者使用过程中取得了一些积极效果,和现有技术相比的确具备很大的优势,下面内容结合试验过程的数据、图表等进行描述。
69.实施例1
70.本发明实施例提供的纯相znsn(oh)6宽带隙金属氢氧化物光催化剂的制备方法,具体包括以下步骤:
71.将1.09g zncl2可溶性盐溶于20ml去离子水中,剧烈搅拌使得zncl2充分完全溶解形成溶液a,浓度为22.7mg/ml;将2.88g na2sno3可溶于48ml去离子水中,剧烈搅拌使得na2sno3充分完全溶解形成溶液b,浓度为60mg/ml;将18ml体积的溶液b逐滴滴入溶液a中,同时进行剧烈搅拌,并将溶液的ph值保持在10左右,形成溶液c;将混合后的反应溶液c倒入聚四氟乙烯水热釜中,盖紧盖子,放入120℃烘箱中,保温24h,反应结束后待降至室温,结束合成过程;将反应完成后的溶液,用去离子水和乙醇洗涤离心5~6次,将最后一次洗涤后的沉淀在60℃的烘箱中干燥10h后,即可得纯相znsn(oh)6宽带隙金属氢氧化物光催化剂。
72.实施例2
73.本发明实施例提供的原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结光催化剂的制备方法,具体包括以下步骤:
74.将1.09g zncl2可溶性盐溶于20ml去离子水中,剧烈搅拌使得zncl2充分完全溶解形成溶液a,浓度为2.7mg/ml;将1.09g na2sno3可溶于48ml去离子水中,剧烈搅拌使得
na2sno3充分完全溶解形成溶液b,浓度为60mg/ml;将24ml体积的溶液b逐滴滴入溶液a中,同时进行剧烈搅拌,并将溶液的ph值保持在10左右,形成溶液c;将混合后的反应溶液c倒入聚四氟乙烯水热釜中,盖紧盖子,放入120℃烘箱中,保温24h,反应结束后待降至室温,结束合成过程;将反应完成后的溶液,用去离子水和乙醇洗涤离心5~6次,将最后一次洗涤后的沉淀在60℃的烘箱中干燥10h后,即可得原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结光催化剂。
75.对实施例1合成的纯相znsn(oh)6宽带隙金属氢氧化物光催化剂和在加入溶液b体积为20ml、22ml、24ml和26ml,水热温度为120℃,水热时间24h所合成原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结光催化剂进行了xrd表征测试(xrd为x-ray diffraction的缩写,即x射线衍射),如图1所示。结果表明,加入溶液b体积为18ml时所合成的光催化剂的xrd谱图与纯相znsn(oh)6相同,未发现sno2相应的峰。说明溶液b体积为18ml时未生成sno2。当加入溶液b体积为20ml以上时发现了微弱的sno2的对应的峰,生成了无定形sno2,但并未改变znsn(oh)6的晶体结构。
76.如图2所示,是实施例1合成的纯相znsn(oh)6宽带隙金属氢氧化物光催化剂,和在加入溶液b体积为24ml和26ml,水热温度为120℃,水热时间24h所合成原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结光催化剂,以及贵金属au(金)和pd(钯)负载的znsn(oh)6光催化剂的xrd图。结果表明,贵金属au(金)和pd(钯)负载在znsn(oh)6表面并未改变znsn(oh)6的晶体结构。
77.如图3所示,是实施例1合成的znsn(oh)6宽带隙金属氢氧化物光催化剂的hrtem图(hrtem为high resolution transmission electron microscope的缩写,即高分辨率透射电子显微镜),从图中可以看到纯相的znsn(oh)6立方体形貌,立方体壁薄,表面光滑。
78.如图4所示,是本发明实施例2添加溶液b体积为24ml,水热温度为120℃,时间为24h,所合成的原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结的tem图和hrtem图(tem为transmission electron microscope的缩写,即透射电子显微镜)。从图中可以看到znsn(oh)6立方体结构,但立方体表面相对粗糙。边缘晶格条纹与无定形sno2匹配,可以得知已经成功的合成了sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结。
79.为了评估上述催化剂对光催化vocs降解和矿化活性,采用连续流的vocs检测系统,并以300w hg灯作为光源来照射光催化剂。在石英玻璃覆盖的流动反应器中评估甲苯矿化的光催化活性。将光催化剂(0.4g)均匀分散在四块石英玻璃板上。将300w高压汞灯放置在反应器顶部约8cm处。在达到吸附-解吸平衡后,在60分钟光照下连续记录甲苯的光催化去除率。冷却装置(包括气体收集装置和风扇)用于将测试系统保持在室温。甲苯气体以100ml/min从气瓶中获得,浓度为500ppm(在n2中)。通过500ml/min的湿空气和440ml/min的干空气混合将相对湿度设置为50%。使用配备光声检测器(gasera one,芬兰)的在线多气体分析仪连续测量甲苯和co2的浓度。甲苯的去除率(η)计算为:η=(1-c/c0)
×
100%,其中c和c0代表出口和进料流的浓度。甲苯的矿化率(ω)计算为:ω=100%
×
(c
’‑c’0)/(c0×
7),其中c'和c'0分别描述为出口流和吸附平衡中的co2浓度。
80.所图5所示,是本发明实施例1合成纯相znsn(oh)6宽带隙金属氢氧化物光催化剂和实施例2在添加溶液b体积为20ml、22ml、24ml和26ml,水热温度为120℃,时间为24h,所合成的原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结光催化剂的光催化vocs
降解和矿化的活性对比图。纯相的znsn(oh)6光催化vocs降解率为5%。随着sno2量的增加,异质结光催化剂的光催化vocs降解率逐渐增加,当b溶液体积为24ml时,zsh24的光催化vocs降解率为72%,是纯相的znsn(oh)6催化活性的15倍。但当继续增加b溶液的体积至26ml,催化活性未进一步提升。
81.为了证明sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结光催化剂表面羟基的作用,在干燥空气中进行光催化vocs降解测试。如图6所示,是实施例2添加溶液b体积为24ml,水热温度为120℃,时间为24h,所合成的原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结在不同湿度条件下的光催化vocs降解和矿化的活性对比图;相较相对湿度50%的反应条件,干燥空气测试条件下的sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结的光催化vocs降解率并未发生改变,表明sno2/znsn(oh)6异质结的表面羟基提供了反应所需羟基。
82.为比较贵金属(金au,钯pd)和sno2对znsn(oh)6表面羟基的活化作用,将表面负载了贵金属au和pd的znsn(oh)6与sno2/znsn(oh)6异质结进行活性对比测试。如图7所示,实施例1合成纯相znsn(oh)6宽带隙金属氢氧化物光催化剂和实施例2添加溶液b体积为24ml,水热温度为120℃,时间为24h,所合成的原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结和贵金属au(金)和pd(钯)负载的znsn(oh)6光催化剂的光催化vocs降解和矿化的活性对比图。结果表明,sno2/znsn(oh)6异质结较au/znsn(oh)6和pd/znsn(oh)6具有更优的催化vocs降解活性。进一步说明,表面原位生成的无定形sno2较贵金属a和pd具有更强的活化表面羟基的能力。
83.为了进一步探究sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结光催化活性提升的原因,选取实施例1合成的纯相znsn(oh)6宽带隙金属氢氧化物光催化剂和实施例2合成的溶液体积为24ml,水热温度为120℃,水热时间24h的sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结进行进一步对比表征,如图8所示。图8是本发明实施例1合成纯相znsn(oh)6宽带隙金属氢氧化物光催化剂和实施例2在添加溶液b体积为24ml,水热温度为120℃,时间为24h,所合成的原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结光催化剂的xps图(xps为x-ray photoelectron spectroscop的缩写,即x射线光电子能谱分析)。从图中发现纯相znsn(oh)6宽带隙金属氢氧化物和sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结的元素xps图谱结合能未发生偏移,说明催化剂中的元素未产生变价,进而说明新生成的sn氧化物是以 4加sn的形态存在,结果表明了sno2的生成。
84.如图9所示,是实施例1合成纯相znsn(oh)6宽带隙金属氢氧化物光催化剂和实施例2在添加溶液b体积为24ml,水热温度为120℃,时间为24h,所合成的原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结光催化剂的uv-vis drs图(uv-vis drs为uv-visible diffuse-reflection spectra,即紫外可见漫反射)。从图中发现,表面无定形sno2的形成,可调节宽带隙金属氢氧化物的带隙宽度,拓宽了纯相znsn(oh)6的光吸收范围,增强了光催化剂的光响应性能。图10是本发明实施例1合成纯相znsn(oh)6宽带隙金属氢氧化物光催化剂和实施例2在添加溶液b体积为24ml,水热温度为120℃,时间为24h,所合成的原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结光催化剂的pl图(pl为photoluminescence的缩写,即荧光光谱)。从pl谱图发现,原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结的荧光强度明显弱于纯相的znsn(oh)6宽带隙金属氢氧化物,
说明原位生成的无定形sno2的存在抑制了电子和空穴的复合。如图11所示,是实施例1合成纯相znsn(oh)6宽带隙金属氢氧化物光催化剂和实施例2在添加溶液b体积为24ml,水热温度为120℃,时间为24h,所合成的原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结光催化剂的光电流强度图。从光电流强度图中可发现,原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结的光电流强度明显强于纯相的znsn(oh)6宽带隙金属氢氧化物,说明原位生成的无定形sno2的存在提高了电荷转移效率,增强了光催化活性。
85.本发明通过在znsn(oh)6宽带隙金属氢氧化物中引入无定形sno2,即保留znsn(oh)6规则的形貌和表面丰富的羟基,又抑制了电子-空穴的复合,提高了光催化vocs降解活性,原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结光催化剂的活性是纯相znsn(oh)6宽带隙金属氢氧化物光催化剂的15.0倍。原位生成sno2/znsn(oh)6宽带隙金属氧化物/氢氧化物异质结光催化剂,未引入新的元素,由一步反应形成,合成简单,反应条件温和。本发明使得宽带隙双金属氢氧化物znsn(oh)6材料具有广泛的应用前景,同时在方法学研究上具有理论意义。
86.以上所述,仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,任何熟悉本技术领域的技术人员在本发明揭露的技术范围内,凡在本发明的精神和原则之内所作的任何修改、等同替换和改进等,都应涵盖在本发明的保护范围之内。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。