一种用于乙烯选择性四聚的催化剂及其应用
1.本技术要求于2021年08月26日提交中国专利局、申请号为202110986355.2、申请名称为“一种用于乙烯选择性四聚的催化剂及其应用”的中国专利申请的优先权,其全部内容通过引用结合在本技术中。
技术领域
2.本发明属于乙烯齐聚催化领域,涉及一种用于乙烯选择性四聚的催化剂及其应用。
背景技术:
3.1-辛烯是重要的化工产品和中间体,主要用于聚乙烯、增塑剂、香精香料、润滑油以及油品添加剂等领域。
4.尽管1-辛烯具有重要的应用价值,但传统的乙烯齐聚技术得到的产物的碳数分布符合schlulz-flory分布,这种分布使齐聚产物中的1-辛烯不可能太高。目前乙烯齐聚合成1-辛烯的方法多是采用铬系催化剂进行催化合成,但该类方法的1-辛烯选择性仍有限,且催化剂成本较高。
技术实现要素:
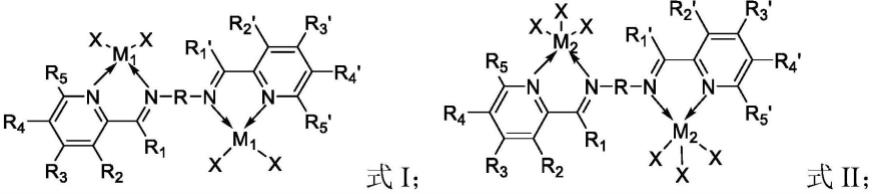
5.本发明提供一种用于乙烯选择性四聚的催化剂,该催化剂中包含式i或式ii所示的双核卤化吡啶亚胺金属配合物,将该催化剂用于乙烯四聚反应中时,反应能够表现出良好的乙烯齐聚活性和高的1-辛烯选择性。
6.本发明还提供一种1-辛烯的制备方法,由于该制备方法以使用如上所述的催化剂催化乙烯齐聚反应,因此该制备方法具有反应活性高、1-辛烯收率高的优点。
7.本发明提供一种用于乙烯选择性四聚的催化剂,包括式i或式ii所示的双核卤化吡啶亚胺金属配合物:
[0008][0009]
式i和式ii中,
[0010]
r独立地选自n=0,1,2,3,4,5,或
[0011]
r1与r1’
各自独立地选自氢、取代或未取代的c1~c6的烷基、取代或未取代的c6~c15的芳基中的一种;
[0012]
r2,r3,r4,r5与r2’
,r3’
,r4’
,r5’
各自独立地选自氢、取代或未取代的c1~c6的烷基、卤素、取代或未取代的c1~c6的烷氧基、取代或未取代的c6~c15的芳基和硝基中的一种;
[0013]
m1选自fe
2
、co
2
和ni
2
中的一种;
[0014]
m2为fe
3
;
[0015]
x独立地选自卤素中的一种。
[0016]
如上所述的催化剂,其中,r1与r1’
各自独立地选自氢、取代或未取代的c1~c4的烷基、取代或未取代的苯基中的一种;
[0017]
和/或,r2,r3,r4,r5与r2’
,r3’
,r4’
r5’
各自独立地选自氢、c1~c4的烷基、氟、氯、溴、甲氧基、乙氧基或硝基中的一种。
[0018]
如上所述的催化剂,其中,r1、r2、r3、r4、r5与r1’
、r2’
、r3’
、r4’
、r5’
中的取代基各自独立地选自氢、烷基、芳基、烷氧基、卤素、硝基中的至少一种。
[0019]
如上所述的催化剂,其中,r选自n=0,2,3。
[0020]
如上所述的催化剂,其中,所述催化剂还包括含铝助催化剂。
[0021]
如上所述的催化剂,其中,所述含铝助催化剂选自铝氧烷和烷基铝化合物中的至少一种。
[0022]
如上所述的催化剂,其中,所述含铝助催化剂中的铝与所述双核卤化吡啶亚胺金属配合物中的金属的摩尔比为(200~2000):1。
[0023]
本发明还提供一种1-辛烯的制备方法,所述制备方法包括:将如上所述的催化剂与有机溶剂混合后,向混合体系通入乙烯进行齐聚反应,得到1-辛烯。
[0024]
如上所述的制备方法,其中,所述有机溶剂选自甲苯、环己烷、乙醚、四氢呋喃、乙醇、苯、二甲苯和二氯甲烷中的至少一种。
[0025]
如上所述的制备方法,其中,式i或式ii所示的双核卤化吡啶亚胺金属配合物在所述混合体系中的摩尔浓度为2~500μmol/l。
[0026]
本发明的催化剂,包含式i或式ii所示的双核卤化吡啶亚胺金属配合物,将该催化剂用于乙烯四聚反应中时,反应能够表现出良好的乙烯齐聚活性和高的1-辛烯选择性。
[0027]
本发明所提供的1-辛烯的制备方法具有反应活性高、1-辛烯收率高的优点。
具体实施方式
[0028]
为使本发明的目的、技术方案和优点更加清楚,下面将结合本发明的实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0029]
本发明第一方面提供一种催化剂,包括式i或式ii所示的双核卤化吡啶亚胺金属配合物:
[0030]
[0031]
式i和式ii中的r可以相同也可以不同,分别可独立地选自n=0,1,2,3,4,5,或
[0032]
式i和式ii中的r1与r1’
可以相同也可以不同,分别可独立地选自氢、取代或未取代的c1~c6的烷基、取代或未取代的c6~c15的芳基中的一种;
[0033]
式i和式ii中的r2,r3,r4,r5与r2’
,r3’
,r4’
,r5’
可以相同也可以不同,分别可独立地选自氢、取代或未取代的c1~c6的烷基、卤素、取代或未取代的c1~c6烷氧基、取代或未取代的c6~c15的芳基和硝基中的一种;
[0034]
式i中的m1选自fe
2
、co
2
和ni
2
中的一种;
[0035]
式ii中的m2为fe
3
;
[0036]
式i和式ii中的x可以相同也可以不同,分别可独立地选自卤素中的一种。
[0037]
在本发明中,术语“c1~c6的烷基”指的是含有1~6个碳原子的饱和直链烃基或饱和支链烃基,具体可以是甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、异戊基、仲戊基、正己基和仲己基等。
[0038]
在本发明中,术语“c1~c6烷氧基”指的是c1~c6的烷基与一个氧原子连接得到的基团,具体可以是甲氧基、乙氧基、正丙氧基、异丙氧基、正丁氧基、异丁氧基、仲丁氧基、叔丁氧基、正戊氧基、仲戊氧基、正己氧基和仲己氧基;特别优选甲氧基和乙氧基等。
[0039]
在本发明中,术语“卤素”指的是氟、氯、溴、碘。
[0040]
在本发明中,术语“c6~c15的芳基”指的是碳原子数为6~15的含芳环的基团,例如可以是苯基、萘基等。
[0041]
发明人研究发现,当催化剂中包括式i或式ii所示的双核卤化吡啶亚胺金属配合物时,该催化剂具有良好的乙烯齐聚活性以及高的1-辛烯选择性。其中,乙烯齐聚活性可达106g/(mol(m)
·h·
atm)以上,1-辛烯选择性可达48.64%以上。
[0042]
进一步的,式i和式ii中的r1与r1’
各自独立地选自氢、取代或未取代的c1~c4的烷基、取代或未取代的苯基中的一种;
[0043]
和/或,r2,r3,r4,r5与r2’
,r3’
,r4’
,r5’
各自独立地选自氢、c1~c4的烷基、取代或未取代的苯基、氟、氯、溴、甲氧基、乙氧基或硝基中的一种。
[0044]
示例性的,c1~c4的烷基可以是甲基、乙基、正丙基、异丙基、丁基、异丁基、仲丁基等。
[0045]
在一种具体的实施方式中,式i或式ii中的r1与r1’
均为甲基,r2,r3,r4,r5与r2’
,r3’
,r4’
r5’
均为氢。
[0046]
在另一种具体的实施方式中,式i或式ii中的r1,r2,r3,r4,r5与r1’
,r2’
,r3’
,r4’
,r5’
均为氢。
[0047]
更进一步的,r1、r2、r3、r4、r5与r1’
、r2’
、r3’
、r4’
、r5’
中的取代基各自独立地选自氢、烷基、芳基、烷氧基、卤素、硝基中的至少一种。此处的取代基是指,当r1、r2、r3、r4、r5与r1’
、r2’
、r3’
、r4’
、r5’
选自被取代基取代的前述基团时,例如取代的c1~c6的烷基、取代的c1~c6烷氧基等,此时取代基选自氢、烷基、芳基、烷氧基、卤素、硝基中的至少一种。其中,烷基可以是c1~c9的直链烷基或支链烷基,如甲基、乙基、丙基、异丙基等,芳基可以是苯基,
萘基等。
[0048]
在一种具体的实施方式中,当r选自n=0,2,3,在使用包括式i或式ii所示的双核卤化吡啶亚胺金属配合物的催化剂进行乙烯齐聚反应时,该反应具有更高的乙烯齐聚活性以及更高的1-辛烯选择性,其中,1-辛烯选择性可高达65%以上。
[0049]
相比于co
2
和ni
2
,当本发明式i所示的双核卤化吡啶亚胺金属配合物中的m1进一步选自fe
2
时,该催化剂表现出更为优异的乙烯齐聚活性和1-辛烯选择性。
[0050]
本发明不限定式i或式ii所示的双核卤化吡啶亚胺金属配合物的制备方法,可采用本领域常规方法制得。
[0051]
在一种具体的实施方法中,式i和式ii所示的双核卤化吡啶亚胺金属配合物可采用下述制备方法制备得到:
[0052][0053]
根据如上反应式所示,使原料a、b、c反应得到配体d,然后将配体d与二卤化金属盐m1x2反应得到式i所示的双核卤化吡啶亚胺金属配合物。同样的,将配体d与三卤化金属盐m2x3反应即可得到式ii所示的双核卤化吡啶亚胺金属配合物。其中,式a中的r1、r2、r3、r4、r5与式i和式ii中的r1、r2、r3、r4、r5中的定义相同,式b中的r1’
、r2’
、r3’
、r4’
、r5’
与式i和式ii中r1’
、r2’
、r3’
、r4’
、r5’
中的定义相同,式c中的r与式i和式ii中r的定义相同。
[0054]
进一步地,本发明的催化剂还包括含铝助催化剂。当催化剂用于乙烯齐聚反应时,含铝助催化剂一方面可消除反应系统中残存的氧分和水分,另一方面可与双核卤化吡啶亚胺金属配合物搭配使用提高催化活性。
[0055]
进一步的,含铝助催化剂选自铝氧烷和烷基铝化合物中的至少一种。
[0056]
具体的,铝氧烷为c1~c4的烷基铝氧烷,进一步优选为甲基铝氧烷、改性甲基铝氧烷、乙基铝氧烷和异丁基铝氧烷,更进一步优选为甲基铝氧烷。
[0057]
具体的,烷基铝化合物的通式为alrnxm,其中,r选自c1~c8的烷基;x为卤素,优选自氯或溴;n与m之和为3,且n为1~3的整数,m为0~2的整数。
[0058]
烷基铝化合物可优选自三甲基铝、三乙基铝、三丙基铝、三异丁基铝、三正己基铝、三正辛基铝、氯化二乙基铝和二氯化乙基铝,更进一步优选为氯化二乙基铝。
[0059]
在一种具体的实施方式中,当含铝助催化剂中的铝与双核卤化吡啶亚胺金属配合物中的金属的摩尔比为(200~2000):1时,催化剂具有更好的催化活性,表现出更为优异的
乙烯齐聚活性和1-辛烯选择性。
[0060]
本发明第二方面提供一种1-辛烯的制备方法,包括:将如上所述的催化剂与有机溶剂混合后,向混合体系通入乙烯进行齐聚反应,得到1-辛烯。
[0061]
如上过程所述的混合是指催化剂的有机溶剂体系和作为反应溶剂的有机溶剂的混合。其中,催化剂的有机溶剂体系可以是催化剂中的各个组分的有机溶剂体系经混合得到,也可以是催化剂中的各个组分混合后再与有机溶剂混合得到。即,上述混合体系是指在通入乙烯前的反应体系。
[0062]
进一步的,上述有机溶剂选自甲苯、环己烷、乙醚、四氢呋喃、乙醇、苯、二甲苯和二氯甲烷中的至少一种。上述有机溶剂均能较好的溶解催化剂,使催化剂在液相状态下进行催化,有利于以更高收率获得1-辛烯。
[0063]
更进一步的,上述有机溶剂可优选自甲苯和二甲苯。
[0064]
可以理解的是,催化剂的浓度也是影响反应速率的反应活性的重要因素,在本发明中,当双核卤化吡啶亚胺金属配合物在混合体系中的摩尔浓度为2~500μmol/l,乙烯在该催化体系中具有良好的齐聚活性以及1-辛烯选择性。
[0065]
在一种具体的实施方式中,在得到混合体系之前,还包括对容置混合体系的反应装置进行预处理。具体的,预处理包括通过高温烘干、真空置换等操作对反应装置进行置换,使反应装置处于无水无氧的状态,然后使用乙烯对反应装置进行置换,使反应装置中充满乙烯。随后,在该预处理的反应装置中完成混合体系和乙烯的齐聚反应。
[0066]
在进行齐聚反应时,较为适宜的反应压力为0.1~30mpa,反应温度为-20~150℃,反应时间为30~100min。
[0067]
本发明的1-辛烯的制备方法,以乙烯为原料,以前述第一方面催化剂催化乙烯的齐聚,最终以较为优异的齐聚活性和α-c8选择性实现了1-辛烯的高效制备。
[0068]
以下,将结合具体的实施例对本发明的催化剂和1-辛烯的制备方法进行说明。
[0069]
下述实施例与对比例中所使用的实验方法如无特殊说明,均为本领域常规方法。下述实施例与对比例中所使用的材料、试剂等如无特殊说明,均可从商业途径得到。
[0070]
实施例1
[0071]
1、催化剂c1的合成
[0072][0073]
1)将吡啶-2-甲醛(2g,18.67mmol)滴加到搅拌的乙醇(30ml)中,再向反应体系中缓慢滴加水合肼(0.37g,wt%=80%,9.33mmol),然后在室温搅拌30分钟沉淀析出,用甲醇(10ml)洗涤,过滤干燥得到黄色固体配体l1(1.5g,7.14mmol,产率76.53%)。
[0074]
配体l1表征如下:
[0075]
lcms 211.2[m 1]
.
[0076]1h nmr(400mhz,dmso-d6)δ8.74(ddd,j=4.8,1.5,0.9hz,2h),8.58(s,2h),8.13(d,j=7.9hz,2h),7.97(td,j=7.7,1.5hz,2h),7.55(ddd,j=7.5,4.8,1.1hz,2h).
[0077]
2)将fecl3(0.65g,0.004mol)溶于乙醇(5ml),然后加入l1(0.42g,0.002mol)的二氯甲烷溶液(5ml),在25℃搅拌12小时沉淀析出,过滤,用乙醚(15ml)洗涤沉淀三次,抽真空干燥得到棕色固体催化剂c1(1.02g,0.0019mol,产率95%)。
[0078]
2、乙烯选择性四聚反应制备1-辛烯
[0079]
1)通过高温烘干、真空置换等操作对反应釜进行置换,确保反应釜中无水无氧;
[0080]
2)使用乙烯继续对反应釜进行置换,使反应釜处于乙烯环境;
[0081]
3)在反应釜中分别加入甲苯溶剂、氯化二乙基铝的甲苯溶液(浓度为1m)和催化剂c1的甲苯溶液,充分搅拌后,通入乙烯开始齐聚反应;
[0082]
其中,反应体系中的溶液总体积为50ml,催化剂c1的浓度为200μmol/l,氯化二乙基铝中的al与主催化剂c1的摩尔比为1000:1;
[0083]
4)使齐聚反应在乙烯压力为1mpa,反应温度为20℃下进行30分钟;
[0084]
5)停止反应,取出少量反应产物用气相色谱进行(gc)分析,通过分析可知,齐聚活性为5.28
×
106g/(mol(fe)
×h×
atm),齐聚物选择性分别为c4 4.59%,c6 1.24%,c8 89.02%(其中含α-c8 73.42%),≥c10 5.15%。除了用于gc分析的少量反应产物外,剩余的混合物用5%的盐酸酸化的乙醇溶液中和,没有得到聚合物。说明本实施例的催化剂不会使乙烯发生聚合生成聚乙烯,避免工业生产中的粘釜现象。
[0085]
上述乙烯齐聚活性以及不同碳数烯烃选择性的计算方法如下:
[0086]
以甲苯溶剂的质量作为参考,根据式1,计算出每个齐聚产物的质量(pi)。
[0087]
通过式2,根据生成的产物量(等于消耗的乙烯量)计算得到乙烯齐聚活性(ao),单位为g/(mol
·h·
atm)。
[0088]
低碳烯烃的选择性(si)是某一低碳烯烃量占总产物量的比例,通过式3计算得到。
[0089][0090]
式1中,ai是指某一齐聚产物的峰面积,a
甲苯
是指甲苯的峰面积,pi是指某一齐聚产物的质量。
[0091][0092]
式2中,ao是指齐聚反应的活性,p1 p2 p3
…
pn是指所有齐聚产物的质量之和,催化剂中金属的物质的量(mol)是指催化剂中金属离子的物质的量,时间(h)是指齐聚反应的时间,压力(atm)是指齐聚反应的压力。
[0093][0094]
式3中,si是指某一齐聚产物的选择性,pi是指某一齐聚产物的质量,p1 p2 p3
…
pn是指所有齐聚产物的质量之和。
[0095]
为方便对比,将分析结果在表1中列出。
[0096]
实施例2
[0097]
1、催化剂c2的合成
[0098][0099]
1)将吡啶-2-甲醛(2g,18.67mmol)滴加到搅拌的乙醇(30ml)中,再向反应体系中缓慢滴加乙二胺(0.56g,9.33mmol),然后加入甲酸(0.01ml),在氮气气氛中加热80℃后搅拌30分钟沉淀析出,将反应液旋干,柱层析分离(洗脱梯度为二氯甲烷:甲醇=50~5:1)得到黄色固体配体l2(1.2g,5.04mmol,产率54.05%)。
[0100]
配体l2表征如下:
[0101]
lcms 239.2[m 1]
.
[0102]1h nmr(400mhz,cdcl3)δ8.61(d,j=4.1hz,2h),8.40(s,2h),7.96(d,j=7.9hz,2h),7.71(td,j=7.7,1.8hz,2h),7.28(ddd,j=7.5,4.9hz,2h),4.05(s,4h).
[0103]
2)催化剂c2的制备步骤与催化剂c1的制备步骤基本一致,不同之处在于,在制备过程中,将实施例1的配体l1替换为本实施例的配体l2。
[0104]
2、本实施例乙烯选择性四聚反应制备1-辛烯的步骤与实施例1基本一致,不同之处在于,将实施例1的催化剂c1替换为本实施例的催化剂c2,气相分析结果见表1。
[0105]
实施例3
[0106]
1、催化剂c3的制备
[0107][0108]
1)将吡啶-2-甲醛(2g,18.67mmol)滴加到搅拌的乙醇(20ml)中,使用乙醇(5ml)将1,3-丙二胺(0.69g,9.33mmol)稀释后缓慢滴加至反应体系中,将反应体系加热至85℃回流10小时。冷却到室温,将反应液旋干得到棕色油状物,将棕色油状物通过柱层析分离(洗脱梯度为二氯甲烷:甲醇=50~5:1)得到黄色固体配体l3(1.1g,4.36mmol,产率46.80%)。
[0109]
配体l3表征如下:
[0110]
lcms 253.1[m 1]
.
[0111]1h nmr(400mhz,cdcl3)δ8.84
–
8.75(m,2h),8.53
–
8.48(m,1h),8.46
–
8.41(m,1h),7.79
–
7.70(m,3h),7.66
–
7.60(m,1h),7.36
–
7.32(m,2h),3.79
–
3.56(m,4h),1.88
–
1.83(m,2h).
[0112]
2)催化剂c3的制备步骤与催化剂c1的制备步骤基本一致,不同之处在于,在制备过程中,将实施例1的配体l1替换为本实施例的配体l3。
[0113]
2、本实施例乙烯选择性四聚反应制备1-辛烯的步骤与实施例1基本一致,不同之处在于,将实施例1的催化剂c1替换为本实施例的催化剂c3,气相分析结果见表1。
[0114]
实施例4
[0115]
1、催化剂c4的制备
[0116][0117]
1)将吡啶-2-甲醛(2g,18.67mmol)滴加到搅拌的乙醇(20ml)中,使用乙醇(5ml)将1,4-二丁胺(0.82g,9.33mmol)稀释后缓慢滴加至反应体系中,将反应体系在25℃反应8小时,有固体析出,过滤,用甲醇洗涤固体(10ml),洗涤后将粗产物使用二氯甲烷和正己烷进行重结晶在二氯甲烷和正己烷得到棕色固体配体l4(1.3g,4.88mmol,产率52.42%)。
[0118]
配体l4表征如下:
[0119]
lcms 267.1[m 1]
.
[0120]1h nmr(400mhz,cdcl3)δppm:8.61(ddd,j=4.8,1.7,0.9hz,2h),8.36(s,2h),7.95(dt,j=7.9,1.0hz,2h),7.73-7.69(m,2h),7.28(ddd,j=7.5,4.9,1.2hz,2h),3.71(td,j=5.2,1.4hz,4h),1.83-1.78(m,4h).
[0121]
2)催化剂c4的制备步骤与催化剂c1的制备步骤基本一致,不同之处在于,在制备过程中,将实施例1的配体l1替换为本实施例的配体l4。
[0122]
3)本实施例乙烯选择性四聚反应制备1-辛烯的步骤与实施例1一致,不同之处在于,将实施例1的催化剂c1替换为本实施例的催化剂c4,气相分析结果见表1。
[0123]
实施例5
[0124]
1、催化剂c5的制备
[0125][0126]
1)将2-乙酰基吡啶(2g,16.51mmol)滴加到搅拌的乙醇(20ml)中,在向反应体系中缓慢滴加水合肼(0.33g,wt%=80%,8.25mmol)和甲酸(0.01ml),然后在25℃搅拌48小时沉淀析出,用甲醇(5ml)洗涤,过滤干燥得到黄色的固体配体l5(1.60g,6.71mmol,产率81.63%)。
[0127]
配体l5表征如下:
[0128]
lcms 239.3[m 1]
.
[0129]1h nmr(400mhz,dmso-d6)δ8.67(ddd,j=4.8,1.6,0.9hz,2h),8.19(d,j=8.0hz,2h),7.91(td,j=7.9,1.8hz,2h),7.49(ddd,j=7.4,4.8,1.1hz,2h),2.30(s,6h).
[0130]
2)催化剂c5的制备步骤与催化剂c1的制备步骤基本一致,不同之处在于,在制备过程中,将实施例1的配体l1替换为本实施例的配体l5。
[0131]
2、本实施例乙烯选择性四聚反应制备1-辛烯的步骤与实施例1基本一致,不同之处在于,将实施例1的催化剂c1替换为本实施例的催化剂c5,气相分析结果见表1。
[0132]
实施例6
[0133]
1、催化剂c6的制备
[0134][0135]
1)将2-乙酰基吡啶(2g,16.51mmol)滴加到搅拌的乙醇(20ml)中,再向反应体系中缓慢滴加乙二胺(0.49g,8.25mmol),然后加入对甲基苯磺酸(0.14g,0.82mmol),在氮气气氛中加热90℃后搅拌12小时沉淀析出,将反应液旋干,柱层析分离(洗脱梯度为二氯甲烷:甲醇=50~5:1)得到黄色固体配体l6(1.3g,4.88mmol,产率59.36%)。
[0136]
配体l6表征如下:
[0137]
lcms 267.4[m 1]
.
[0138]1hnmr(400mhz,cdcl3)δ8.58(ddd,j=4.8,1.8,0.9hz,2h),8.06(dt,j=8.0,1.0hz,2h),7.71-7.66(m,2h),7.26(ddd,j=7.4,4.8,1.2hz,2h),3.96(s,4h),2.42(s,6h).
[0139]
2)催化剂c6的制备步骤与催化剂c1的制备步骤基本一致,不同之处在于,在制备过程中,将实施例1的配体l1替换为本实施例的配体l6。
[0140]
2、本实施例乙烯选择性四聚反应制备1-辛烯的步骤与实施例1基本一致,不同之处在于,将实施例1的催化剂c1替换为本实施例的催化剂c6,气相分析结果见表1。
[0141]
实施例7
[0142]
1、催化剂c7的制备
[0143][0144]
1)将2-乙酰基吡啶(2g,16.51mmol)滴加到搅拌的乙醇(20ml)中,再向反应体系中缓慢滴加丙二胺(0.61g,8.25mmol),然后加入对甲基苯磺酸(0.14g,0.82mmol),在氮气气氛中加热90℃搅拌12小时冷却至室温沉淀析出,过滤干燥得到黄色固体配体l7(1.2g,4.28mmol,产率51.85%)。
[0145]
配体l7表征如下:
[0146]
lcms 281.1[m 1]
.
[0147]1h nmr(400mhz,cdcl3)δ8.60-8.53(m,2h),7.92-7.68(m,4h),7.60(ddd,j=8.6,7.4,1.8hz,2h),3.70-3.53(m,4h),2.34-2.23(m,6h),1.96-1.91(m,2h).
[0148]
2)催化剂c7的制备步骤与催化剂c1的制备步骤基本一致,不同之处在于,在制备过程中,将实施例1的配体l1替换为本实施例的配体l7。
[0149]
2、本实施例乙烯选择性四聚反应制备1-辛烯的步骤与实施例1基本一致,不同之处在于,将实施例1的催化剂c1替换为本实施例的催化剂c7,气相分析结果见表1。
[0150]
实施例8
[0151]
1、催化剂c8的制备
[0152][0153]
1)将2-乙酰基吡啶(2g,16.51mmol)滴加到搅拌的乙醇(20ml)中,再向反应体系中缓慢滴加丁二胺(0.73g,8.25mmol),然后加入对甲基苯磺酸(0.14g,0.82mmol),在氮气气氛中加热90℃搅拌12小时,旋干反应液,柱层析分离提纯(洗脱梯度为二氯甲烷:甲醇=50~5:1)得到黄色固体配体l8(1.3g,4.42mmol,产率56.52%)。
[0154]
配体l8表征如下:
[0155]
lcms 295.3[m 1]
.
[0156]1h nmr(400mhz,cdcl3)δ8.61
–
8.56(m,2h),7.85
–
7.81(m,2h),7.72(ddd,j=8.2,7.5,1.5hz,2h),7.61(ddd,j=8.5,7.5,1.5hz,2h),3.79
–
3.66(m,4h),2.44
–
2.24(m,6h),1.81
–
1.71(m,4h).
[0157]
2)催化剂c8的制备步骤与催化剂c1的制备步骤基本一致,不同之处在于,在制备过程中,将实施例1的配体l1替换为本实施例的配体l8。
[0158]
2、本实施例乙烯选择性四聚反应制备1-辛烯的步骤与实施例1基本一致,不同之处在于,将实施例1的催化剂c1替换为本实施例的催化剂c8,气相分析结果见表1。
[0159]
实施例9
[0160]
1、催化剂c9的制备
[0161][0162]
1)将2-乙酰基吡啶(2g,16.51mmol)滴加到搅拌的甲苯(20ml)中,再向反应体系中加入1,4-苯二胺(1.01g,9.34mmol),使用甲苯(5ml)将对甲基苯磺酸(0.16g,0.93mmol)溶解后滴入反应液中,将反应液在100℃氮气气氛中回流10小时。将反应液冷却到室温,有固体析出,用正己烷洗涤得到黄色固体配体l10(1.5g,5.24mmol,产率56.18%)。
[0163]
2)催化剂c9的制备步骤与催化剂c1的制备步骤基本一致,不同之处在于,在制备过程中,将实施例1的配体l1替换为本实施例的配体l9。
[0164]
2、本实施例乙烯选择性四聚反应制备1-辛烯的步骤与实施例1基本一致,不同之处在于,将实施例1的催化剂c1替换为本实施例的催化剂c9,气相分析结果见表1。
[0165]
实施例10
[0166]
本实施例与实施例6基本一致,不同之处在于,在乙烯选择性四聚反应制备1-辛烯的过程中,保持乙烯压力为2mpa。
[0167]
本实施例气相分析结果见表1。
[0168]
实施例11
[0169]
1、催化剂c10的制备
[0170][0171]
1)本实施例催化剂配体l6的制备与实施例6一致;
[0172]
2)将fecl2·
2h2o(0.65g,0.004mol)和配体l6(0.53g,0.002mol)溶于乙醇和二氯甲烷的混合溶剂(10ml)中,将反应混合物在25℃搅拌5小时沉淀析出,过滤,用乙醚(15ml)洗涤沉淀三次,在真空烘箱中60℃干燥得到黄色固体催化剂c10。
[0173]
2、本实施例乙烯选择性四聚反应制备1-辛烯的步骤与实施例1基本一致,不同之处在于,将实施例1的催化剂c1替换为本实施例的催化剂c10,气相分析结果见表1。
[0174]
实施例12
[0175]
1、催化剂c11的制备
[0176][0177]
1)本实施例催化剂配体l6的制备与实施例6一致;
[0178]
2)将cocl2·
6h2o(0.95g,0.004mol)和l6(0.53g,0.002mol)溶于乙醇和二氯甲烷的混合溶剂(10ml)中,将反应混合物在25℃搅拌5小时沉淀析出,过滤,用乙醚(15ml)洗涤沉淀三次,在真空烘箱中60℃干燥得到黄色固体催化剂c11。
[0179]
2、本实施例乙烯选择性四聚反应制备1-辛烯的步骤与实施例1基本一致,不同之处在于,将实施例1的催化剂c1替换为本实施例的催化剂c11,气相分析结果见表1。
[0180]
实施例13
[0181]
1、催化剂c12的制备
[0182][0183]
1)本实施例催化剂配体l6的制备与实施例6一致;
[0184]
2)将nicl2·
6h2o(0.95g,0.004mol)和l6(0.53g,0.002mol)溶于乙醇和二氯甲烷的混合溶剂(10ml)中,将反应混合物在25℃搅拌5小时沉淀析出,过滤,用乙醚(15ml)洗涤沉淀三次,在真空烘箱中60℃干燥得到黄色固体催化剂c12。
[0185]
2、本实施例乙烯选择性四聚反应制备1-辛烯的步骤与实施例1基本一致,不同之处在于,将实施例1的催化剂c1替换为本实施例的催化剂c12,气相分析结果见表1。
[0186]
实施例14
[0187]
本实施例与实施例1基本一致,不同之处在于,改变氯化二乙基铝的用量,使氯化二乙基铝中的al与助催化剂c1的摩尔比为500:1。本实施例气相分析结果见表1。
[0188]
实施例15
[0189]
本实施例与实施例1基本一致,不同之处在于,氯化二乙基铝在乙烯选择性四聚反应制备1-辛烯的过程中,将助催化剂氯化二乙基铝替换为甲基铝氧烷。本实施例气相分析结果见表1。
[0190]
表1
[0191][0192][0193]
通过表1的数据可以看出,将本发明的催化剂用于乙烯四聚反应中,反应主要生成为c8产物,其中,c8产物中α-c8(1-辛烯)为优势产物,从而说明本发明的催化剂用于乙烯四聚反应时具有良好的齐聚活性以及1-辛烯选择性。
[0194]
最后应说明的是:以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。