扩增或者检测含有全长poly(a)的全长转录组cdna的方法
技术领域
1.本发明属于生物检测领域,具体涉及扩增或者检测含有全长poly(a)的全长转录组cdna的方法及其应用。
背景技术:
2.全长转录组测序作为生命科学研究的基础,促进了各物种基因功能、基因表达调控以及进化关系等多方面的基础与应用研究。目前全长转录组的研究实际上是只包含部分poly(a)的读取,无法反映poly(a)长度的真实信息,而rna poly(a)尾巴是成熟的mrna和lncrna的重要组成部分,对rna稳定性和翻译也有重要的调控作用,现poly(a)尾检测的相关技术仍然非常有限,二代测序无法对同聚物长序列进行读取。近期,文献报道了一种新的基于pacbio平台的poly(a)长度的检测方法paiso-seq,可在获得全长转录组信息的同时得到poly(a)长度信息。
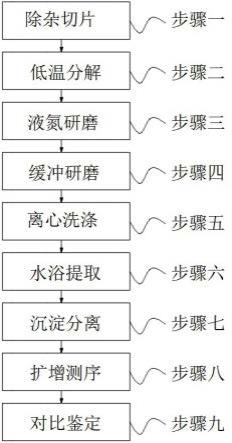
3.目前基于pacbio iso-seq开发了一套可检测的poly(a)尾检测技术paiso-seq,实现了对rna poly(a)尾序列的测定。建库过程:首先通过含u的oligo dt进行末端延伸,延伸完后将含u的oligo dt进行消化,保留rt引物进行模板置换,获得全长cdna并进行扩增再进行环状接头连接构建了保留rna poly(a)尾的paiso-seq文库,进行测序。(详细建库流程见:https://www.nature.com/articles/s41467-019-13228-9#sec11的fig.1。文献出处:liu,y.,nie,h.,liu,h.et al.poly(a)inclusive rna isoform sequencing(paiso-seq)reveals wide-spread non-adenosine residues within rna poly(a)tails.nat commun 10,5292(2019).https://doi.org/10.1038/s41467-019-13228-9。)
4.现有技术主要面向群体细胞水平,而在单细胞水平目前受限于方法的检测通量,无法实现高通量单细胞含全长poly(a)的转录组检测,单细胞全长转录本(含全长poly(a))导致检测时间长,成本高。
技术实现要素:
5.本发明所要解决的技术问题是为克服现有技术中不能高通量地检测或者扩增包含全长poly(a)长度的转录组,只能挑单个细胞进行反应的缺陷,提供一种扩增或者检测含有全长poly(a)的全长转录组cdna的方法。本发明的所述方法可以实现单细胞层面的包含全长poly(a)长度的全长转录组检测。且基于10x genomics和pacbio平台的高通量单细胞的全长poly(a)转录组产品,提供了更精细化演化细胞发育或改变的过程的完整方案。
6.一方面,尽管目前高通量技术发展相对较为成熟,然而尚没有高通量是针对可以检测polya的技术。且目前的检测带上polyt,也只是为了捕捉到mrna,通过随机的结合在polya的任意位置来检测到mrna的信息。但本发明人特异性地利用10x genomics平台中gel beads上的关键性结构,调整了原10x的试剂盒内的master mix使其不局限于cdna合成,并结合tso技术检测,能够实现高通量单细胞的全长poly(a)转录组的检测,提供了更精细化演化细胞发育或改变的过程的完整方案。
7.另一方面,目前的高通量(同时标记上千个单细胞以便区分)的单细胞测序需要依托于微流控技术将单个细胞分离,且高通量的单细胞测序只能检测3端序列;而能检测polya全长的技术无法做到高通量,因此本技术在不变更现有元件的情况下可以做含polya全长的转录组检测,有其不可替代的作用。
8.本发明通过以下技术方案解决上述技术问题。
9.本发明的技术方案之一为:一种基于10x genomics单细胞平台扩增或者检测含有全长poly(a)的全长转录组cdna的方法,所述方法包括:
10.(1)单细胞反应制备;其包括a)制备单细胞悬液;b)配制master mix;c)将a)和b)混匀,得混匀液并制备液滴;其中所述master mix包括dntp、无5
’‑
》3’外切酶活性的dna聚合酶和缓冲液;
11.(2)末端延伸:以带poly(dt)的10
×
barcoded gel beads为模板,使带poly(a)的转录本末端延伸得单链的产物1;
12.(3)模板置换:对所述产物1进行反转录,在反转录产物后的3’末端添加3个碱基c;并添加tso引物进行反应得产物2;
13.(4)pcr扩增:对所述产物2进行pcr扩增。
14.所述步骤(1)中用到的缓冲液可为常规的逆转录缓冲液。例如为可包含50mm tris-hcl、ph 8.3,75mm kcl,3mm mgcl2以及0.02m dtt的缓冲液;也可以为neb缓冲液2,例如包含50mm nacl,10mm tris-hcl,10mm mgcl2和1mm dtt的缓冲液。
15.在本发明一较佳实施方案中,以上步骤(1)的“a)制备单细胞悬浮液”中,每43.2μl所述单细胞悬浮液通过将8μl rnase抑制剂以及浓度为700~1200cells/μl的标准品细胞混合,并补足nf水制得;所述标准品细胞的个数优选10000个。
16.在本发明一较佳实施方案中,以上步骤(1)的“b)配制master mix”中,每31.8μl所述master mix包含6.1μl浓度为10μm的dntp、9.7μl浓度为50units/μl的klenow片段、14μl 10
×
neb缓冲液2以及2μl还原剂b;所述缓冲液为10
×
neb缓冲液2。
17.在本发明一较佳实施方案中,以上步骤(1)的“c)所述制备液滴”为:将所述混匀液、gel beads和partitioning oil分别依次加入到gem chip g的row labeled 1、2和3中,在gem chip g上覆盖上10
×
gasket并制备液滴。
18.优选地,在所述液滴中,所述单细胞悬浮液、master mix、gel beads和partitioning oil的体积比为43.2:31.8:50:45。
19.较佳地,步骤(2)所述的末端延伸包括:
20.a)将步骤(1)中所得产物于37℃反应1小时;
21.b)向步骤a)所得产物按4:5体积比加入recovery agent,待液体分层后去除recovery agent与partitioning oil的混合液体;其中a)中所得产物使用例如100μl,b)中所述recovery agent使用例如125μl。
22.此外,在步骤(3)之前优选对步骤(2)所得产物进行样品纯化和引物消化。
23.其中,所述样品纯化较佳地包括:
24.1)将dynabeads myone silane、cleanup buffer、reducing agent b与nuclease-free water混匀,配制dynabeads cleanup mix;其中,所述dynabeads myone silane、所述cleanup buffer、所述reducing agent b以及nuclease-free water的体积比例如为8:
182:5:5,所述dynabeads cleanup mix的体积例如为200μl;
25.2)将所述dynabeads cleanup mix与步骤(2)所得产物混匀、室温静置;可选地,再吹打混匀一次室温静置;所述静置的时间例如为5分钟;
26.3)按dynabeads cleanup mix和80%乙醇为2:3的体积比在2)中加入80%乙醇,管内静置,弃上清;所述静置的时间优选30秒;
27.4)可选地,重复步骤3)一次,将弃上清后的离心管短离2s,静置至澄清,去掉残留的乙醇;
28.5)加入elution solution i回溶;所述elution solution i与步骤3)所得混合液的体积比例如为1:10~1:15;较佳地,所述elution solution i通过将buffer eb、10%tween 20以及reducing agent b混合后制备得到,所述buffer eb、所述10%tween 20以及所述reducing agent b的体积比优选98:1:1;所述reducing agent b例如来自10x genomics;
29.6)吹打混匀后放于室温25℃静置10min。
30.关于引物消化,现有技术中通常通过在primer上设计加上u,再用user酶消化含有u的primer再将消化下来的primer用磁珠纯化掉。本发明中可直接利用外切酶i(exonuclease i)消化掉未参与的单链的primer再纯化。
31.本发明中所述引物消化较佳地包括:
32.1)在纯化产物管内加入等量exonuclease i和rnase抑制剂,再加入10
×
neb buffer 2放置在37℃反应;所述反应时间优选30min,所述纯化产物、所述exonuclease i、所述rnase抑制剂以及所述10x neb buffer 2的体积比例如为(30~35):1:1:4;
33.2)取1.8倍体积的室温rnaclean xp beads加入pcr产物中并充分混匀,室温孵育;所述孵育时间例如为5min;
34.3)静置例如2-5min至液体澄清,弃掉上清;加入约等倍体积的80%乙醇,室温静置例如30s后弃去上清;
35.4)重复步骤3),弃上清并将管底液体尽量吸干;
36.5)室温干燥,使磁珠表面无反光、无开裂;
37.6)加入te进行cdna洗脱,移液器吹打混匀并室温下溶解;所述溶解的时间优选5min;
38.7)将离心管置于磁力架上,静置至液体澄清,将上清液转移到新离心管中;所述液体澄清的时间例如为2-5min。
39.步骤(3)中较佳地使用superscripttm ii/iv进行所述反转录。
40.在本发明一较佳实施方案中,步骤(4)中每50μl所述pcr扩增的体系包括:
41.9μl步骤(3)所得产物、25μl 2x的kapa hifi hotstart readymix、2μl浓度为10μm的如seq id no:1所示的上游引物、2μl浓度为10μm的如seq id no:2所示的下游引物以及12μl nf水。
42.本发明的技术方案之二为:一种含poly(a)的全长转录组的建库方法,所述建库方法包括如技术方案之一所述的方法。
43.基于技术方案之一所述的方法,所述建库方法较佳地还包括对所述步骤(4)所得产物进行末端修复。
44.所述末端修复的反应体系的体积较佳地为30μl;更佳地,每30μl所述反应体系包含:25μl所述产物,3.5μl nebnext μltra ii end prep reaction buffer、1.5μl nebnext μltra ii end prep enzyme mix。
45.所述末端修复的反应条件较佳地为:将所述反应体系于20℃反应30min后,于65℃反应30min。
46.进行所述末端修复后还可对产物进行接头的添加,所述接头的序列例如如seq id no:3所示。
47.添加接头后优选还进行酶消化反应和磁珠纯化;
48.较佳地,所述酶消化的反应体系中,每95μl含有42.5μl加接头后产物、10μl反应缓冲液、1μl浓度为20u/μl的核酸外切酶i、1μl浓度为100u/μl的核酸外切酶iii以及40.5μl的无核酸酶水。
49.在符合本领域常识的基础上,上述各优选条件,可任意组合,即得本发明各较佳实例。
50.本发明所用试剂和原料均市售可得。
51.本发明的积极进步效果在于:
52.本发明可以通过末端延伸,单细胞平台在微滴环境内对单细胞转录组进行延伸完后将单细胞转录组标记上分子标签,保留rt引物进行模板置换,获得全长cdna并进行扩增再进行环状接头连接构建了保留rna poly(a)尾的完整转录组文库,本发明提高了对单细胞全长转录组的建库通量,弥补了目前单细胞转录组检测不能检测全长poly(a)的技术空白。
附图说明
53.图1为高通量单细胞含poly(a)的全长转录组建库技术原理。
54.图2为含poly(a)的全长转录组建库文库质检结果。
55.图3为poly(a)长度分布。
具体实施方式
56.下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
57.实施例1使用本发明的一种高通量单细胞含poly(a)的全长转录组建库技术对乳癌细胞系4t1细胞进行建库测序
58.使用的试剂盒及试剂主要包括chromium next gem chip g single cell kit,16rxns pn-1000127(10x genomics,1000127)、chromium next gem single cell 3
′
gem,library&gel bead kit v3.1(10x genomics,1000128)、qubit
tm rna hs assay kit(invitrogen)、dsdna br assay kit or hs assay kit(invitrogen)、2100high sensitivity dna kit(agilent)、pb beads(pacific biosciences)。
59.建库原理以及过程详见图1。具体如下:
60.1.单细胞poly(a)末端延伸
61.1.1单细胞反应制备
62.1)根据实验需要取已计数浓度为700~1200(cells/μl)的标准品细胞10000个至一个标记好的0.2ml pcr管里,加8μl rnase inhibitor(invitrogen)再补nf水至体积43.2μl配制好细胞悬浮液。
63.2)取另一个0.2ml pcr管标记好为master mix,然后配制反应体系见下表1:
64.表1
65.试剂名称单管用量(μl)dntp(10μm,neb)6.1klenow fragment(50units/μl neb)9.7neb buffer 2(10x,neb)14reducing agent b(10x genomics)2total31.8
66.3)将细胞悬浮液与master mix轻柔混匀后,取70μl混匀液加入至gem chip g的row labeled 1中。
67.4)准备gel beads,从-80℃取出后,待融化后再震荡混匀30秒,短暂离心5秒,缓慢吸取50μl加入到gem chip g的row labeled 2中。
68.5)取45μl partitioning oil,gem chip g的row labeled 3。在gem chip g上覆盖上10x gasket,再放入到controller制备微滴,时长约18min;
69.6)微滴生成后取出微滴生成液约100μl转至一个标记好的0.2ml pcr管里,放置冰上待用。
70.1.2末端延伸
71.1)将上一步的产物(100μl)放入pcr仪,37℃反应1小时。
72.2)反应完毕后加入125μl recovery agent到样本中等待2min,待液体分层后缓慢移除掉125μl的recovery agent与partitioning oil混合液体(粉色)。
73.1.3样品纯化
74.1)从4℃冰箱取出dynabeads myone silane(10x genomics)并充分混匀后取8μl至新的250μlep管内,再在管内加入cleanup buffer(10xgenomics)182μl、reducing agent b(10x genomics)5μl以及nuclease-free water 5μl配制共200μl的dynabeads cleanup mix。
75.2)将配制好的dynabeads cleanup mix200μl加入1.2(3)的产物中混匀室温静置5分钟,再吹打混匀一次再室温静置5分钟后,放置磁力架上。
76.3)使用300μl的新鲜配制的80%乙醇,管内等待30秒,弃上清。
77.4)重复上一步骤一次,将去上清后的离心管短离2s,放于磁力架上静止至澄清,去掉残留的乙醇。
78.5)配制elution solution i:buffer eb 98μl 10%tween 20(现配)1μl reducing agent b(10x genomics)1μl,共100μl。
79.6)开盖晾干1min后,用34.5μl elution solution i回溶,吹打混匀后放于室温25℃静置10min。
80.7)10min结束后,取出250μl离心管,短离2s,放置于磁力架上,静止至澄清,吸取上
清液,回收于另一个标记好的250μl ep离心管中。
81.1.4引物消化
82.1)在纯化产物管内加入exonuclease i(e.coli,neb)和rnase抑制剂(invitrogen)各1μl,再加入neb buffer 2(10x,neb)4μl放置在37℃反应30min;
83.2)提前30min取出rnaclean xp beads(beckman coμlter,inc)置于室温,使用前充分震荡混匀;取1.8倍体积rnaclean xp beads加入pcr产物中,移液器轻轻吹打至少10次充分混匀,室温孵育5min;
84.3)将离心管置于磁力架上,静置2-5min至液体澄清,移液器吸取并弃掉上清;保持离心管固定于磁力架上,加入120μl新鲜配制的80%乙醇,室温静置30s后小心弃去上清;
85.4)重复上一步骤一次,弃上清后顺利后放置磁力架,将管底液体尽量吸干;
86.注意:不要吸取磁珠,以免影响产量。
87.5)保持离心管固定于磁力架上,打开离心管管盖,室温干燥,直至磁珠表面无反光、无开裂;
88.6)将离心管从磁力架上取下,加入8.5μl te进行cdna洗脱,移液器吹打混匀并室温下溶解5min;
89.7)将离心管置于磁力架上,静置2-5min至液体澄清,将上清液转移到新离心管中。
90.2.模板置换
91.2.1cdna合成(反转录rt)
92.1)取纯化后产物(8μl)已延伸rna进行rt。反应体系及反应条件见下表2:
93.表2
[0094][0095][0096]
其中rt primer序列为5
’‑
ctacacgacgctcttccgatct-3’(seq id no:4)。
[0097]
2)完成反应后cdna用0.8x ampure pb磁珠纯化。
[0098]
3.pcr扩增
[0099]
1)在上一步的cdna纯化回收10μl,配制pcr体系,反应体系及反应条件见下表3:
[0100]
表3
[0101][0102][0103]
其中,pcr primer f序列为5
’‑
ctacacgacgctcttccgatct-3’(seq id no:1)
[0104]
pcr primer r序列为5
’‑
aagcagtggtatcaacgcagag-3’(seq id no:2)
[0105]
2)反应结束后,将pcr产物转至1.5ml离心管中,进行0.8x ampure pb磁珠纯化。
[0106]
4.smrtbell
tm
文库构建
[0107]
4.1末端修复及加接头
[0108]
1)使用nebnextμltra ii dna library prep kit for illumina试剂盒进行,末端修复体系见下表:
[0109]
表4
[0110][0111]
2)完成末端修复后进行加接头,反应体系见下表:
[0112]
表5
[0113][0114]
/5phos/nnnnnnnnnnnnnnnn(16bpbarcode)
[0115]
atctctctcttttcctcctcctccgttgttgttgttgagagagatnnnnnnnnnnnnnnnn((16bpbarcode)t(seq id no:3)
[0116]
4.2酶消化反应及磁珠纯化
[0117]
1)向上步产物中加入表6所示混合液。详细反应体系见下表:
[0118]
表6
[0119][0120][0121]
2)反应完成后,取新的1.5ml离心管,做好标记,将反应后体系全部转移到新的对应的离心管中。
[0122]
3)取0.5倍体积(50μl)的ampure pb磁珠加入到装有到酶消化反应产物的1.5ml离心管中,进行纯化,最终回溶于50μl洗脱缓冲液中。
[0123]
4)取1μl纯化好的样品稀释5倍,取1μl稀释的样品进行qubit检测,并将剩余的稀释样品进行2100质检。
[0124]
5.上机测序
[0125]
在建库后均采用pacbio公司提供的loading方案,按procedure&checklist
–
preparing single-cell isoseq
tm libraries using express templateprep kit 2.0说明书要求进行制备上机并测序。
[0126]
6.测试结果
[0127]
使用4t1细胞系进行含poly(a)的全长转录组建库上机。
[0128]
1)文库出库质检结果:
[0129]
建库结束后,对文库进行2100质检,质检结果显示文库主峰均在1.7k左右,均处于
正常范围内。见图2。
[0130]
含poly(a)的全长转录组建库文库质检结果
[0131]
2)建库方法所得测序数据检测poly(a)的长度见下表以及图3。
[0132]
根据图3可知:本发明方法测到polya尾的长度分布从30nt至400多nt左右,平均polya尾长度为83.21nt。
[0133]
表7
[0134]
文库平均poly(a)长度含poly(a)的全长转录组83.21nt
再多了解一些
本文用于创业者技术爱好者查询,仅供学习研究,如用于商业用途,请联系技术所有人。