1.本发明涉及一种能够用于在温和的条件下合成氨的复合氧化物,使用该复合氧化物的金属担载物和氨合成催化剂,以及该复合氧化物的制造方法、该金属担载物的制造方法和氨的制造方法。
背景技术:
2.氨是现代的化学产业中的重要原料。所生产的氨的80%以上,被用来制造农作物用的化学肥料。另外,氨作为能量和氢的载体也引人注目。这是由于:(1)其氢含有量较多(17.6wt%),(2)能量密度高(12.8gj/m3),(3)为了制造氢而进行分解时不会产生二氧化碳。如果能够由太阳能、风能等可再生能源高效率地制造氨,那么能够减轻能源以及粮食危机所相关的地球规模的问题。
3.目前,用于制造氨的哈勃博斯(haber-bosch)法,会大量消耗能量,其量占世界的能量消耗量的约1~2%。在该方法中,消耗能量的约60%被回收,可确保氨的焓值。但是,剩余的能量的大部分,在通过天然气制造氢时、合成氨时、以及分离气体时损耗。由于哈勃博斯法的氨合成在非常高的温度(>450℃)和压力(>20mpa)下进行,因此亟需减少在该方法中使用的大量的能量。为了抑制地球规模的能量消耗量,需要与哈勃博斯法中使用的铁基体的催化剂相比,能够在更加温和的条件(更低的温度和压力)合成氨的催化剂。
4.近年,已知在1mpa(10个气压)左右的低压条件下制造氨的方法。制造氨所使用的钌催化剂,通常被担载在载体上。例如,在专利文献1中公开了,若使用稀土类氧化物作为担载钌的载体,则能够降低钌的使用量,并且能够降低反应温度。但是,在专利文献1的氨制造方法中,在较低压条件下制造氨时,氨收获率不够。因此,本发明人,开发了一种将在650℃下被还原的la
0.5
ce
0.5o1.75
用作载体的钌催化剂,并报告其在低压条件下也示出了优良的特性(非专利文献4)。
5.进一步,本发明人开发了由2种金属元素形成的二元系的复合氧化物,和在该复合氧化物上担载钌等催化剂的金属担载物(氨合成用催化剂)(专利文献5、专利文献6)。作为在本文献中公开的使用了二元系的复合氧化物(载体)的氨合成用催化剂,公开了如下催化剂。
6.ru/ce
0.85
la
0.15ox
_(500℃、600℃、650℃、700℃)还原,ru/ce
0.67
la
0.33ox
_(500℃、600℃、650℃、700℃)还原,ru/ce
0.33
la
0.67ox
_(500℃、600℃、650℃、700℃)还原,ru/ce
0.15
la
0.85ox
_(500℃、600℃、650℃、700℃)还原,ru/ce
0.5
la
0.5ox
_(500℃、650℃、800℃)还原,ru/ce
0.5
zr
0.5ox
_700℃还原,ru/ce
0.5
pr
0.5ox
_(500℃、600℃、650℃、700℃,800℃)还原,ru/la
0.5
pr
0.5ox
_(450℃、500℃、600℃、650℃、700℃)还原,ru/ba
0.1
la
0.9ox
_(500℃、700℃、800℃、900℃)还原,ru/ba
0.1
ce
0.9ox
_(500℃、700℃)还原,co/ba
0.05
la
0.95ox
_(500℃、600℃、700℃、800℃)还原,co/ba
0.01
la
0.99ox
_700℃还原,co/ba
0.03
la
0.97ox
_700℃还原,co/ba
0.1
la
0.9ox
_700℃还原。
7.进一步,本文献中还记载了8.4wt%ba/4.5wt%ru/mgo_(500℃,700℃)还原(实
施例80、实施例81)。这些氧化物,是使得作为载体的mgo含浸ru溶液后进行烧制,进一步,使用ba(oh)2·
8h2o以使其担载ba而得到的氧化物。
8.除了专利文献1以及非专利文献4以外,在各样的专利文献中公开了在各种各样的稀土类氧化物载体上担载钌的氨合成催化剂。作为代表,可列举专利文献2~4、非专利文献1~3。在专利文献2和专利文献4中公开了选用镧系元素氧化物作为载体,在专利文献3中公开了选用氧化镨作为载体,在非专利文献1中公开了选用ce氧化物作为载体。在非专利文献2中公开了,将ru、ce、la的氢氧化物共同沉淀、干燥、活性化而制造的ru/ceo2-la2o3系的催化剂。
9.在包括专利文献1、2、4和非专利文献1的现有技术的文献中记载了,在合成氨所使用的钌催化剂中,ru以粒子的形式存在于其载体表面。在以粒子的形式存在的情况下,有其平均直径大于5nm的报道(参照非专利文献2),并且有小于2nm的报道(非专利文献4)。另外,在专利文献3中,公开了ru为蛋壳结构。
10.另一方面,关于载体,非专利文献3中公开了,在评价担载有ru的y(la)-m-o(m为ca、sr、ba)催化剂的氨合成活性时,对于担载ru之前的载体氧化物,发现将载体氧化物的烧结温度设为450℃时载体氧化物的比表面积增大,将烧结温度提高到650℃时载体的比表面积减小。
11.另外,鉴于ru的价格昂贵,还提出了在载体上担载除了ru以外的过渡金属化合物,例如担载co的氨合成用催化剂(例如,参照非专利文献5、非专利文献6)。然而,在非专利文献6中,虽然公开了在钡氧化物上担载钴的co-bao/c,但是氨合成活性低。另外,非专利文献5中,虽然未使用氧化物而使用氨基钙(co/ba-ca(nh2)2)),但是该担载有co的催化剂的1mpa下的氨收获率,不及担载ru的催化剂。
12.现有技术文献
13.专利文献
14.专利文献1:日本特开平6-079177号公报
15.专利文献2:日本特开2013-111562号公报
16.专利文献3:国际公开第2016/133213号
17.专利文献4:日本特开2017-018907号公报
18.专利文献5:国际公开第2019/059190号
19.专利文献6:国际公开第2019/216304号
20.非专利文献
21.非专利文献1:y.niwa and k.aika,chemistry letters,(1996)3-4
22.非专利文献2:x.luo et al.,catalysis letters 133,382(2009)
23.非专利文献3:a.s.ivanova et al.,kinetics and catalysis,vol.45,no.4,2004,pp.541-546.translated from kinetika i kataliz,vol.45,no.4,2004,pp.574-579.
24.非专利文献4:y.ogura et al.,「efficient ammonia synthesis over a ru/la
0,5
ce
0.5o1.75 catalyst pre-reduced at high temperature」,chemical science,第9卷,2230~2237页
25.非专利文献5:m.kitano et al.,angew.chem.int.ed.,130(2018)2678
26.非专利文献6:w.gao et.al.,acs catal.,7(2017)3654
技术实现要素:
27.发明要解决的技术问题
28.合成用催化剂通常需要较高的合成活性。对于目前正在开发的氨合成用的催化剂,持续一直需求能够得到更高的收获率的高活性的催化剂。在二元系的催化剂中,例如co/balao
x
这样的催化剂,虽然氨合成活性足够高,但是需要进一步提高活性。
29.本发明的目的在于,提供一种在担载例如钴时可示出比现有的含有稀土类的二元系的复合氧化物例如balao
x
更高的氨合成活性的复合氧化物。另外,本发明的另一目的在于,提供一种示出如此高的氨合成活性的金属担载物以及氨合成用催化剂。进一步,本发明的又一目的在于,提供一种这样的复合氧化物和金属担载物的制造方法以及氨的制造方法。
30.解决技术问题的方法
31.本发明人为了解决上述技术问题而发现,作为构成复合氧化物的金属氧化物,通过组合使用具有特定的性质的2种第2族元素,在制成催化剂时氨合成活性高,从而得到以下的发明。
32.〔1〕一种复合氧化物,是由金属元素l的氧化物和金属元素n的氧化物形成的复合氧化物,其特征在于,用下述通式(1)的组成表示,并且满足以下(a)~(d):
33.lnn
1-n
ꢀꢀꢀ
(1)
34.(a)所述金属元素l,是从以下的(i)~(iii)的任一者中选择的元素的氧化物:
35.(i)第1族元素,
36.(ii)第2族元素,
37.(iii)第1族元素和第2族元素;
38.(b)所述金属元素n,由除了金属元素l以外的第1族或第2族元素组成;
39.(c)所述n在0.001以上0.300以下;
40.(d)所述金属元素l的氧化物与金属元素n的氧化物没有形成固溶体,而是所述金属元素l的氧化物粒子堆积于所述金属元素n的氧化物粒子的表面。
41.〔2〕如〔1〕所述的复合氧化物,其中,
42.(a)所述金属元素l,表示氧化物的状态下的氧的部分负电荷(-δ
oa
)的值在0.56以上0.70以下的作为强碱性元素的金属元素,
43.(b)所述金属元素n,表示氧化物的状态下的氧的部分负电荷(-δ
ob
)的值在0.35以上0.55以下的作为弱碱性元素的金属元素。
44.〔3〕如〔1〕或〔2〕所述的复合氧化物,其特征在于,是由金属元素l中所包括的金属元素a与金属元素n中所包括的金属元素b构成的二元系的复合氧化物,所述通式(1)用下述通式(2)的组成表示,并且满足以下的(a)~(d):
45.a
nb1-n
ꢀꢀꢀ
(2)
46.(a)所述金属元素a,表示氧化物的状态下的氧的部分负电荷(-δ
oa
)的值在0.56以上0.70以下的作为强碱性元素的第2族元素;
47.(b)所述金属元素b,表示氧化物的状态下的氧的部分负电荷(-δ
ob
)的值在0.35以
上0.55以下的作为弱碱性元素的第2族元素;
48.(c)所述n在0.001以上0.300以下;
49.(d)所述金属元素a的氧化物与所述金属元素b的氧化物没有形成固溶体,而是所述金属元素a的氧化物粒子堆积于所述金属元素b的氧化物粒子的表面。
50.〔4〕一种复合氧化物,是由金属元素l的氧化物和金属元素n的氧化物形成的复合氧化物,其特征在于,用下述通式(3)的组成表示,并且满足以下(a)~(d):
51.lnn
1-nox
ꢀꢀꢀ
(3)
52.(a)所述金属元素l,是从以下的(i)~(iii)的任一者中选择的元素的氧化物:
53.(i)第1族元素,
54.(ii)第2族元素,
55.(iii)第1族元素和第2族元素,
56.(b)所述金属元素n,由除了金属元素l以外的第1族元素或第2族元素构成;
57.(c)所述n在0.001以上0.300以下;
58.(d)所述金属元素l的氧化物与所述金属元素n的氧化物没有形成固溶体,而是所述金属元素l的氧化物粒子堆积于所述金属元素n的氧化物粒子的表面。
59.〔5〕如〔4〕所述的复合氧化物,其中,
60.(a)所述金属元素l,表示氧化物的状态下的氧的部分负电荷(-δ
oa
)的值在0.56以上0.70以下的作为强碱性元素的金属元素,
61.(b)所述金属元素n,表示氧化物的状态下的氧的部分负电荷(-δ
ob
)的值在0.35以上0.55以下的作为弱碱性元素的金属元素。
62.〔6〕如〔4〕或〔5〕所述的复合氧化物,其特征在于,是由金属元素l中所包括的金属元素a与金属元素n中所包括的金属元素b构成的二元系的复合氧化物,所述通式(3)用下述通式(4)的组成表示,并且满足以下的(a)~(d):
63.a
nb1-nox
ꢀꢀꢀ
(4)
64.(a)所述金属元素a,表示氧化物的状态下的氧的部分负电荷(-δ
oa
)的值在0.56以上0.70以下的作为强碱性元素的第2族元素;
65.(b)所述金属元素b,表示氧化物的状态下的氧的部分负电荷(-δ
ob
)的值在0.35以上0.55以下的作为弱碱性元素的第2族元素;
66.(c)所述n在0.001以上0.300以下;
67.(d)所述金属元素a的氧化物与所述金属元素b的氧化物没有形成固溶体,而是所述金属元素a的氧化物粒子堆积于所述金属元素b的氧化物粒子的表面;
68.(e)所述x表示复合氧化物保持电中性所需要的氧原子的个数。
69.〔7〕如〔1〕~〔6〕中任一项所述的复合氧化物,其特征在于,复合氧化物是banmg
1-nox
,其中,0.001≤n≤0.300。
70.〔8〕如〔7〕所述的复合氧化物,其特征在于,复合氧化物是banmg
1-nox
,其中,0.01≤n≤0.10。
71.〔9〕如〔7〕或〔8〕所述的复合氧化物,其特征在于,所述复合氧化物中含有的碳酸盐的量,与ba相比为10mol%以下。
72.〔10〕一种金属担载物,其特征在于,在如〔1〕~〔9〕中任一项所述的复合氧化物上,
担载从钴、铁、镍所构成的群组中选择的至少1种金属粒子m。
73.〔10-1〕一种金属担载物,是由所述金属粒子m和所述〔1〕~〔3〕中任一项所述的复合氧化物形成、并且可确认各种元素为粒子的集合体的组合物,其中,可观察到粒径为所述金属粒子m的粒径的10%以下的金属元素l,分布在所述金属粒子m的粒子上、分布在所述金属粒子m与所述金属氧化物n之间、以及分布在所述金属氧化物n上。
74.〔10-2〕如〔10-1〕所述的金属担载物,其中,可确认金属元素l粒子的分布是均匀地分布。
75.〔10-3〕如〔10-1〕或「10-2]所述的金属担载物,其中,所述金属元素l的粒子,还分布在金属粒子m与金属元素n的中间层。
76.〔10-4〕如〔10-1〕~〔10-3〕中任一项所述的金属担载物,其中,所述复合氧化物是如〔6〕所述的复合氧化物,金属元素a选自金属元素l,金属元素b选自金属元素n。
77.〔11〕如〔10〕所述的金属担载物,其中,在堆积于金属元素n的氧化物的表面的金属元素l的氧化物上担载所述金属粒子m,并且在金属粒子m的表面上堆积有金属元素l的氧化物粒子。
78.〔12〕如〔10〕所述的金属担载物,其中,在所述金属元素l的氧化物粒子与所述金属粒子m之间分布有所述金属元素n的氧化物粒子。
79.〔12-1〕如〔11〕或〔12〕所述的金属担载物,其中,所述复合氧化物是如〔6〕所述的复合氧化物,金属元素a选自金属元素l,金属元素b选自金属元素n。
80.〔13〕如〔10〕所述的金属担载物,其特征在于,所述金属粒子m是钴粒子。
81.〔14〕一种氨合成用催化剂,其特征在于,使用了如〔10〕所述的金属担载物。
82.〔15〕一种如所述〔10〕所述的金属担载物的制造方法,其中,包括下述(a)~(d)的步骤:
83.(a)浸渍步骤,使包含所述金属元素n的n前体含浸包含所述金属元素l的l前体;
84.(b)复合氧化物烧制步骤,在500℃以上的温度下对所述混合物进行烧制得到由复合氧化物构成的载体;
85.(c)担载步骤,让所述复合氧化物含浸包含所述金属粒子m的化合物的前体得到含浸载体;和
86.(d)担载物烧制步骤,在400℃以上的温度下对所述含浸载体进行烧制。
87.〔15―1〕如所述〔15〕所述的金属担载物的制造方法,其中,金属元素a选自金属元素l,金属元素b选自金属元素n,所述方法包括下述(a)~(d)的步骤:
88.(a)浸渍步骤,使包含所述金属元素b的b前体含浸包含所述金属元素a的a前体溶液;
89.(b)复合氧化物烧制步骤,在500℃以上的温度下对所述混合物进行烧制得到由复合氧化物构成的载体;
90.(c)担载步骤,使所述复合氧化物含浸包含所述金属粒子m的化合物的前体溶液而得到含浸载体;和
91.(d)担载物烧制步骤,在400℃以上的温度下对所述含浸载体进行烧制。
92.〔15-2〕如〔15〕或〔15-1〕所述的金属担载物的制造方法,其特征在于,在空气中进行所述(b)步骤。
93.〔15-3〕如〔15〕或〔15-1〕所述的金属担载物的制造方法,其特征在于,在氩气中进行所述(d)步骤。
94.〔16〕如〔15〕~〔15-3〕中任一项所述的金属担载物的制造方法,其特征在于,还包括步骤(e):
95.(e)还原步骤,将通过所述(d)得到的金属担载物在氢的存在下、在500℃以上进行烧制。
96.〔17〕一种氨的制造方法,其是使得氢和氮与催化剂接触而制造氨的方法,其特征在于,所述催化剂是如〔14〕所述的氨合成用催化剂。
97.发明的效果
98.根据本发明,能够提供一种复合氧化物,其在担载例如钴时,与现有的包含稀土类的二元系的复合氧化物、例如ru/balao
x
相比,可示出更高的氨合成活性。另外,根据本发明,能够提供一种示出如此高的氨合成活性的金属担载物以及氨合成用催化剂。进一步,根据本发明,能够提供一种这样的复合氧化物和金属担载物的制造方法,以及氨的制造方法。
附图说明
99.图1是示出实施例中的co/ba
0.01
mg
0.99ox
_700℃还原催化剂(担载co后在700℃下还原而配制的催化剂)的氨合成活性的图表。
100.图2是示出使用在实施例的co/bamgo
x
催化剂中改变向载体添加的ba的量而配制的co/bamgo
x
催化剂来测定的氨合成活性的图表。
101.图3是示出使用在实施例的co/bamgo
x
催化剂中改变向载体添加的ba的量而配制的bamgo
x
催化剂来测定的氨合成活性的图表。
102.图4是示出使用在实施例的co/bamgo
x
催化剂中改变还原温度而配制的co/bamgo
x
催化剂来测定的氨合成活性的图表。
103.图5是示出使用在实施例的co/bamgo
x
催化剂中改变还原温度而配制的co/bamgo
x
催化剂来测定的氨合成活性的图表。
104.图6是使用在实施例的co/bamgo
x
催化剂中改变还原温度而配制的co/bamgo
x
催化剂来测定的xrd图谱。
105.图7是示出使用在实施例的co/bamgo
x
催化剂中改变co担载量而配制的co/bamgo
x
催化剂来测定的氨合成活性的图表。
106.图8是示出使用在实施例的co/bamgo
x
催化剂中改变co担载量而配制的co/bamgo
x
催化剂来测定的氨合成活性的图表。
107.图9是示出在实施例的co/bamgo
x
催化剂中改变反应压力来测定的氨合成活性的图表。
108.图10是示出在实施例的co/bamgo
x
催化剂中改变反应压力来测定的氨合成活性的图表。
109.图11是示出在实施例的co/bamgo
x
催化剂中改变反应压力来测定的氨合成活性的图表。
110.图12是示出使用在实施例的co/bamgo
x
催化剂中改变前处理条件而配制的co/bamgo
x
催化剂来测定的氨合成活性的图表。
111.图13是示出使用在实施例的co/bamgo
x
催化剂中改变co前体而配制的co/bamgo
x
催化剂来测定的氨合成活性的图表。
112.图14是使用在实施例的co/bamgo
x
催化剂中改变co前体而配制的co/bamgo
x
催化剂来测定的xrd图谱。
113.图15是示出在实施例的co/bamgo
x
催化剂中改变sv来测定的氨合成活性的图表。
114.图16是示出在实施例的co/bamgo
x
催化剂中改变sv来测定的氨合成活性的图表。
115.图17是示出在实施例的co/bamgo
x
催化剂中将反应温度设为低温来测定的氨合成活性的图表。
116.图18是示出在实施例的co/bamgo
x
催化剂中将反应温度设为低温来测定的氨合成活性的图表。
117.图19是示出实施例的ru/bamgo
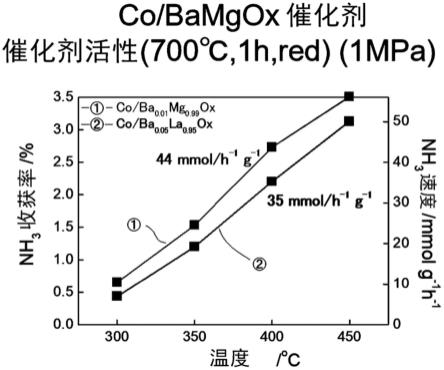
x
的氨合成活性的图表。
118.图20是示出实施例中的fe/ba
0.01
mg
0.99ox
_700℃还原催化剂(担载co后在700℃下还原而配制的催化剂)的氨合成活性的图表。
119.图21是示出使用对实施例的fe/bamgo
x
催化剂的前处理条件进行改变而配制的co/bamgo
x
催化剂所测定的氨合成活性的图表。
120.图22是示出实施例的co/bamgo
x
催化剂中的co-fe催化剂的氨合成活性的图表。
121.图23是实施例1的co/bamgo
x
催化剂的h2-tpr测定结果。
122.图24是比较例2的co/mgo
x
催化剂的h2-tpr测定结果。
123.图25是表示实施例1的co/bamgo
x
催化剂的通过tem得到的元素分布的图。分别为:图25(1)是haadf-stem像,图25(2)是ba的元素分布图,图25(3)是mg的元素分布图,图25(4)是co的元素分布图,图25(5)是ba、co、mg的元素分布叠加的图。
124.图26是示出将实施例的co/bamgo
x
催化剂中的ba改变为其他的第2族元素得到的催化剂的氨合成活性的图表。
125.图27是示出使用第1族元素代替第2族元素配制的催化剂来测定的氨合成活性的图表。
126.图28是示出使用添加第2族元素和第1族元素双方而配制的载体得到的催化剂的氨合成活性的图表。
127.图29是示出使用添加第2族元素和第1族元素双方而配制的载体得到的催化剂的氨合成活性的图表。
128.图30是示出使用添加第2族元素和第1族元素双方而配制的载体得到的催化剂的氨合成活性的图表。
129.图31是示出使用添加第2族元素和第1族元素双方而配制的载体得到的催化剂的氨合成活性的图表。
130.图32是示出使用利用了各种过渡金属元素而配制的载体得到的催化剂的氨合成活性的图表。
131.图33是示出使用利用了各种过渡金属元素而配制的载体得到的催化剂的氨合成活性的图表。
132.图34是表示实施例17的co/bamgo
x
催化剂的通过tem得到的元素分布的图。分别为:图34(1)是haadf-stem像,图34(2)是ba的元素分布图,图34(3)是mg的元素分布图,图
34(4)是co的元素分布图,图34(5)是ba、co、mg的元素图叠加的图。
133.图35是对实施例1、17的金属担载物的结构的特征进行说明的图。
134.图36是标准化的催化剂以及标准试料的xanes光谱。
具体实施方式
135.<复合氧化物>
136.本发明的复合氧化物,是由金属元素l的氧化物和金属元素n的氧化物形成的复合氧化物,其特征在于,用下述通式(1)的组成表示,并且满足以下(a)~(d):
137.lnn
1-n
ꢀꢀꢀ
(1)
138.其中,金属元素l,是从以下的(i)~(iii)的任一者中选择的元素的氧化物:
139.(i)第1族元素,
140.(ii)第2族元素,
141.(iii)第1族元素和第2族元素。
142.需要说明的是,在本发明中“金属元素l”,不仅包括1种元素(第1族元素或第2族元素),也包括2种元素(第1族元素和第2族元素)。
143.作为金属元素l的第1族元素,是被称作所谓的碱金属的金属,可列举li、na、k、rb、cs。另外,第2族元素,是被称作碱土类金属的金属,可列举be、mg、ca、sr、ba、ra等。另外,存在同时使用第1族元素和第2族元素作为金属元素l的情况。关于这些元素的选择,可在考虑碱性的基础上进行选择。进一步,也能够计算下文所述的部分负电荷并进行使用。在使用第1族金属和第2族金属的情况下,其比率优选在0.1:1.9~1.9:0.1的范围内,更优选在0.8:1.2~1.2:0.8的范围内,特别优选为1:1。基于下文所述的金属担载物的氨合成活性较高的点,作为金属元素l,优选是单独的第2族元素、或者第1族元素和第2族元素的复合氧化物。
144.金属元素n,可使用除了金属元素元素l以外的第1族或第2族元素的氧化物。金属元素l相对于金属元素n使用量少。与金属元素l的金属元素总量相比的金属元素n,通常以0.001以上且0.300以下进行使用,优选地,以0.01以上且0.100以下进行使用。金属元素l的比率,会影响催化剂制造时的形态,也会给催化剂活性带来影响。金属元素l的氧化物与金属元素n的氧化物没有形成固溶体,在金属元素n上观察到了金属元素l。在本说明书中,有时也将该状态称为:金属元素l或金属元素l的氧化物,堆积在金属元素n或金属元素n的氧化物上。
145.本发明中使用的复合氧化物,金属元素a选自金属元素l,金属元素b选自金属元素l,在为二元系的复合氧化物的情况下,通式(1)用下述通式(2)表示。
146.a
nb1-n
ꢀꢀꢀ
(2)
147.(a)所述金属元素a,是作为第2族元素的碱土类金属,并且是氧化物的状态下的氧的部分负电荷(-δ
oa
)的值为0.56以上0.70以下的强碱性元素。
148.(b)所述金属元素b,是除了金属元素a以外的碱土类金属,并且表示氧化物的状态下的氧的部分负电荷(-δ
ob
)的值为0.35以上0.55以下的作为弱碱性元素的第2族元素。
149.(c)与整体相比金属元素a的添加比率,为0.001以上0.300以下,该范围的情况下的形态会给催化剂活性带来正面影响。
150.(d)另外,将所述金属元素a的氧化物与所述金属元素b的氧化物没有形成固溶体,
而在所述金属元素b的氧化物粒子的表面上观察到所述金属元素a的氧化物粒子的状态,在本说明书称为“堆积”。
151.如下所示,通式(1)的组成表示的复合氧化物,如果用其他的表述记载,则能够写成式(3)。即,本发明中使用的复合氧化物,是由金属元素l的氧化物和金属元素n的氧化物形成的复合氧化物,其特征在于,用下述通式(3)的组成表示,并且满足以下(a)~(d):
152.lnn
1-nox
ꢀꢀꢀ
(3)
153.(a)金属元素l,是从以下的(i)~(iii)的任一者中选择的元素的氧化物:
154.(i)第1族元素,
155.(ii)第2族元素,
156.(iii)第1族元素和第2族元素;
157.(b)金属元素n,由除了金属元素l以外的第1族元素或第2族元素构成;
158.(c)所述n在0.001以上0.300以下;
159.(d)所述金属元素l的氧化物与所述金属元素n的氧化物没有形成固溶体,而是所述金属元素l的氧化物粒子堆积于所述金属元素n的氧化物粒子的表面;
160.(e)所述x表示复合氧化物保持电中性所需要的氧原子的个数。
161.本发明的复合氧化物,如果将上述通式(2)用其他方式表述,则是用下述通式(4)的组成表示的、由金属元素a以及金属元素b形成的二元系的复合氧化物。
162.a
nb1-nox
ꢀꢀꢀ
(4)
163.(其中,a、b、n、x如在上述通式(2)中所说明的。)
164.<碱性>
165.本发明中使用的复合氧化物,金属元素l的氧化物和金属元素n的氧化物没有形成固溶体。所述金属元素l,优选是氧化物的状态下的氧的部分负电荷(-δ
oa
)的值为0.56以上0.70以下的作为强碱性元素的金属元素,所述金属元素n,优选是氧化物的状态下的氧的部分负电荷(-δ
ob
)的值为0.35以上0.55以下的作为弱碱性元素的金属元素。
166.在金属元素a选自金属元素l,且金属元素b选自金属元素n,并形成二元系的复合氧化物的情况下:
167.(a)所述金属元素a,表示氧化物的状态下的氧的部分负电荷(-δ
oa
)的值为0.56以上0.70以下的作为强碱性元素的第2族元素,
168.(b)所述金属元素b,表示氧化物的状态下的氧的部分负电荷(-δ
ob
)的值为0.35以上0.55以下的作为弱碱性元素的第2族元素,
169.(c)所述n在0.001以上0.300以下,
170.(d)所述金属元素a的氧化物与所述金属元素b的氧化物没有形成固溶体,而是所述金属元素a的氧化物粒子堆积于所述金属元素b的氧化物粒子的表面,
171.(e)所述x表示复合氧化物保持电中性所需要的氧原子的个数。
172.金属元素a,是氧化物的状态下的氧的部分负电荷(-δo)的值在0.56以上0.70以下的强碱性元素。-δ
oa
的值更优选为0.60以上,最优选为0.65以上。具体地,金属元素a能够从ba(钡)、sr(锶)、ca(钙)中选择。
173.金属元素b,是氧化物的状态下的氧的部分负电荷(-δo)的值在0.35以上0.55以下的弱碱性元素。-δ
ob
的值更优选为0.40以上。另外,-δ
ob
的值更优选为0.50以下,最优选
为0.45以下。具体地,金属元素b,能够从mg(镁)、be(铍)中选择。
174.-δ
oa
与-δ
ob
的差,优选为0.10~0.40,更优选为0.15~0.35,最优选为0.20~0.30。
175.在本发明中,复合氧化物中包含的金属元素a,是在氧化物的状态下示出高的碱性的强碱性元素,因此能够提高氨合成催化剂的活性。以下,对其原理的概要进行说明。
176.金属元素a是强碱性的金属元素。从所述元素的复合氧化物(载体)的碱基点产生电子,该电子通过担载于该复合氧化物的作为催化剂的过渡金属粒子被反而供应给氮分子,减弱氮三键。发明人,将该阶段称作氨合成反应的控速阶段,可认为通过上述的一系列的电子的动作,降低了切断氮分子的三键的能量,提高了金属担载物(催化剂)的氨合成活性。
177.金属氧化物的碱性(路易斯碱性)与电子提供能力的高低相关。即,可认为电子提供能力越高的物质示出越强的碱性。基本上在氧化物中,氧作为电子提供体发挥作用,因此氧化物中的氧所带有的电荷的量,即氧的部分负电荷能够用作碱性的指标。实际上,在非专利文献(桑德森《无机化学(上)》广川书店(1975年)第276页表12.7)中示出,氧的部分负电荷的值与氧化物所示出的酸碱性强烈相关。
178.这里,关于各别的金属元素的氧化物,氧的部分负电荷(-δo),能够使用在非专利文献(桑德森《无机化学(上)》广川书店(1975年)第276页)的表12.7中记载的值,没有记载值的氧化物,能够通过上述的氧的部分负电荷的计算而算出。在下述表中,示出了第2族元素的氧化物的氧的部分负电荷(-δo)的值。
179.【表1】
[0180] beomgocaosrobao氧的部分负电荷0.350.420.570.620.67碱性vw(微弱碱性)w(弱碱性)s(强碱性)s(强碱性)s(强碱性)酸性vw(微酸性)o(无酸性)o(无酸性)0(无酸性)o(无酸性)
[0181]
另一方面,关于复合氧化物整体,氧的部分负电荷的计算方法,参照非专利文献(桑德森《无机化学(上)》广川书店(1975年)第122页表6.7,126~128页)。首先,求出复合氧化物中的各元素的组成比。例如,“ce
0.5
la
0.5o1.75”的la为0.5,将该值记做ni(i为对应的元素)。另外,将各种元素的电负性度记做χi。然后,通过(π(χi
ni
))^(1/σni)求出构成复合氧化物的全部原子的电负性度的几何平均。接着,为了求出氧的电负性度的变化,求出所述几何平均与氧的电负性度(5.21)之差。最后,将氧的电负性度的变化,除以每1个氧原子得到1个电子的情况下的电负性度的变化(-4.75)。通过以上的计算,能够计算出复合氧化物所示出的氧的部分负电荷。
[0182]
总结上文,关于复合氧化物的氧的部分负电荷的值,当该复合氧化物中包含的各元素的组成比记做ni(i表示至少包含a、b、o的复合氧化物中的全部元素),并将各元素的电负性度记做χi(i表示至少包含a、b、o的复合氧化物中的全部元素)时,用下式(a)表示:
[0183]
((π(χi
ni
))^(1/∑ni)-5.21)/-4.75
··
式(a)。
[0184]
在计算复合氧化物的氧的部分负电荷的情况下,形成复合体的元素中,(a)可以通过处于氧化物的状态的氧的部分负电荷求出,(b)当将复合氧化物中包含的各元素的组成比记做ni(i表示包含a、b、o的复合氧化物中的全部元素),并将各元素的桑德森电负性度记
做χi(i表示包含a、b、o的复合氧化物中的全部元素)时,可以通过下述式(a)求出:
[0185]
((π(χi
ni
))^(1/σni)―5.21)/-4.75
··
式(a)。
[0186]
在复合氧化物形成均匀的复合氧化物的情况下,优选进行(b)的方法。另一方面,在复合氧化物形成不均匀的复合氧化物的情况下,优选进行(a)的方法,在该情况下可以使用单个元素的氧的部分负电荷中绝对值最大的元素的结果。在本发明的复合氧化物中,由于金属元素a的氧化物和金属元素b的氧化物没有形成固溶体而形成分相,因此复合氧化物的氧的部分负电荷的值,优选通过上述的(a)的方法进行计算。
[0187]
复合氧化物的氧的部分负电荷的值,优选为0.35以上,更优选为0.40以上。当复合氧化物的氧的部分负电荷的值为0.35以上时,存在氨合成活性变高的倾向。
[0188]
<碳酸盐、氢氧化物的除去>
[0189]
金属元素l(碱金属)、n(碱土类金属),即便是氧化物碱性也很强,容易与大气中的二氧化碳和水反应而成为金属碳酸盐和氢氧化物。但是,该金属碳酸盐以及氢氧化物会降低复合氧化物的碱性,并成为催化剂的氨合成活性降低的原因。例如,ba在大气中容易形成为baco3、ba(oh)2,这会降低氨合成活性。因此,氨合成催化剂中含有的金属碳酸盐和氢氧化物尽可能地越少越好。为了减少金属碳酸盐和氢氧化物,优选在如下文所述的加热条件下进行还原处理,由此将催化剂中含有的金属碳酸盐和氢氧化物分解,能够防止碱性降低。金属担载物中包含的碳酸盐的量,只要在不会妨碍氨合成活性的范围内就没有特别的限制,例如,与金属元素a相比为10摩尔%以下,优选为1摩尔%以下,更优选为0.1摩尔%以下,还更优选为0.01摩尔%以下。
[0190]
作为对以金属碳酸盐存在的碳酸盐的量进行定量的方法,可列举在氢流通下加热催化剂,从而使碳酸种氢化而产生甲烷等烃,并用质量分析计或氢火焰离子化检测器(fid)、热传导检测器(tcd)等检测出烃,再进行换算的方法。
[0191]
另外,也能够使用对金属的碳酸盐的敏感度高的红外吸收分光法。对催化剂照射红外光,并测定碳酸盐特征性地吸收的波数的峰的吸收强度,从而能够对催化剂中含有的碳酸盐的量进行定量。例如,碳酸ba的定量所能够使用的峰的位置,在3000cm
-1
附近、2450cm
-1
附近、1750cm
-1
附近、1480cm
-1
附近、1060cm
-1
附近等。
[0192]
下述表中示出了第2族元素中的氧的部分负电荷的值较大的ba、sr,和作为碱金属的cs、k的氧化物等的融点。
[0193]
【表2】
[0194][0195]
根据下文所述的h2-tpr测定结果,可认为发生了以下的反应。
[0196]
baco3 4h2→
bao ch4 2h2o
ꢀꢀꢀ
(5)
[0197]
另外,虽然ba的氧化物的融点高,但是如上所述,在氢气气氛下的加热处理时,会中间生成融点较低的氢氧化物。因此,通过氢氧化物的融解得到流动性。此时可认为:由于ba和金属元素b均作为强碱性的化合物而存在,因此相互排斥,或者由于界面张力、又或由于其他的某种理由,所以结果是ba的化合物向金属粒子上流动,生成ba的氢氧化物以粒子状分布于金属粒子的表面的状态,在反应后又成为氧化物而丧失流动性同时体积收缩,从而在具有空隙的该状态下直接被固定化,因而发现了较高的活性。在本发明中,将金属表面上还有粒子状存在的状态称作“分布”、“堆积”的状态。
[0198]
如此,元素本来的碱性强,并且减少对其产生阻碍的碳酸盐也很容易,因此基于能够提高氨合成活性的点,作为金属元素a特别优选ba。
[0199]
金属元素b的氧化物,是复合氧化物的主要成分,在本发明中示出了弱碱性(也包括微弱碱性。在以下的说明书中同样如此。)。现有技术文献中记载的作为二元系的复合氧化物的banla
1-nox
中,ba和la这2种金属元素的氧化物的部分负电荷的值都较高(ba为0.67,la为0.56),与之相反,在发明中,金属元素b的氧化物的部分负电荷的值较低为0.35以上0.55以下。
[0200]
<堆积>
[0201]
在本发明中,金属元素l的氧化物和金属元素n的氧化物没有形成固溶体而形成分相,金属元素l的氧化物堆积于金属元素n的氧化物粒子的表面,并且堆积对金属元素l的氧化物进行担载的金属粒子的表面。因此,即使金属元素n的氧化物的碱性较弱,只要金属元素l的氧化物的碱性强,就可通过金属元素l的氧化物来提高氨合成活性。因此,即使金属元素n的氧化物比与现有的低,也可示出较高的氨合成活性。
[0202]
作为金属元素n的氧化物,优选比表面积(ssa)较大。这是由于,作为复合氧化物的主要成分的金属元素n的氧化物的比表面积较大时,能够牢固地固定co等微细的纳米粒子,并且这些纳米粒子的活性部位的数量增多,而氨合成活性提高。因此,基于比表面积的大小的观点,作为金属元素n,特别优选mg(镁)。虽然作为复合氧化物金属元素n是主要成分,但
是在用作担载过渡金属的担载物以形成催化剂的情况下,金属元素n的氧化物发挥了很大的作为载体的作用。
[0203]
基于上述观点,作为式(1)和式(2)的复合氧化物,优选为banmg
1-nox
(其中,0.001≤n≤0.300)。
[0204]
这里,关于banmg
1-nox
,ba的组成比(即,n的值),优选在0.01≤n≤0.10的范围内。如下文所述的实施例所示,当在0.01≤n≤0.10的范围内时,有氨合成活性(收获率、收获量)增高的倾向。
[0205]
<金属担载物>
[0206]
本发明的金属担载物,是在本发明的复合氧化物上担载除了第4族之外的过渡金属的粒子(以下,有时称作过渡金属粒子)的金属担载物。作为过渡金属,基于催化剂活性高的观点,优选从ru、fe、co、ni、rh、pd、os、ir、pt所构成的群组中选择1种以上,更优选为ru、co或fe与co的混合物。其中,基于与本发明的复合氧化物组合时的氨合成活性的高度,特别优选co。过渡金属与复合氧化物的量比,能够考虑催化剂活性与过渡金属的成本进行确定,例如,优选过渡金属与金属担载物整体相比的比例处于0.1~50重量%的范围,更优选处于5.0~30重量%的范围。
[0207]
本发明的过渡金属m,与金属元素l的氧化物和金属元素n的氧化物中任一者均没有形成固溶体。特别优选为,金属元素l的氧化物粒子堆积并且覆盖在过渡金属m的粒子上的结构体,进一步堆积在作为主要成分的金属元素n的氧化物粒子的表面上。即,优选处于以过渡金属m为核、且以金属元素n为壳的所谓的核/壳的关系。过渡金属m的整体被金属元素l的氧化物粒子覆盖,并且隔着金属元素l的氧化物被担载在金属元素n的氧化物粒子上。可认为,这样的结构是,在金属n上担载金属l,之后担载过渡金属m,可认为是为了在进行高温还原处理的时候让具有流动性的金属l的氧化物覆盖过渡金属m。当金属l的量与作为载体的金属元素n的量相比过多,或者,与过渡金属的量相比过多时,层会过厚,因此氮和氢无法到达作为活性点的过渡金属表面,这是不优选的。另外,当在700℃以上的高温或者在进行长时间的烧制时,金属l的氧化物粒子会凝集,因而氮和氢无法到达作为活性点的过渡金属表面,因此是不优选的。
[0208]
本发明的复合氧化物,是金属元素l的氧化物和金属元素n的氧化物的混合状态。特别地,优选是在作为主要成分的金属元素n的氧化物粒子的表面上,堆积有金属元素l的氧化物粒子的层叠结构。需要说明的是,最优选的是,两者处于所谓的核/壳的关系。进一步,催化剂载体内部不存在金属元素l也可以,优选金属元素l存在于载体表面。
[0209]
特别地,本发明的复合氧化物,是金属元素a的氧化物和金属元素b的氧化物的混合状态。特别地,优选是在作为主要成分的金属元素b的氧化物粒子的表面上,堆积有金属元素a的氧化物粒子的层叠结构。需要说明的是,最优选的是,两者处于所谓的核/壳的关系。进一步,催化剂载体内部不存在金属元素a也可,优选金属元素a存在于载体表面。
[0210]
由于是金属元素l的氧化物与金属元素n的氧化物没有固溶而处于混合状态(分相),因此在用作下文所述的金属担载物(催化剂)时,过渡金属粒子在复合氧化物的表面与金属元素l的氧化物直接接触。由于金属元素l(例如ba)的氧化物是强碱性,所以可推测,因为过渡金属粒子与这些氧化物进进行直接接触所以示出高活性的活性点增多,氨合成活性提高。另一方面,在co的情况下,阳离子的碱性很重要,ba等金属元素a的氧化物的氧的部分
负电荷越大,则氨合成活性越高。
[0211]
金属元素a的氧化物粒子与金属粒子m的粒径比a/m,通常为20%以下,优选为10%以下,更优选为5%以下。为了改变该粒径,可对金属元素a与过渡金属m之比进行调节。当该粒径比过大或者过小时,有难以得到所预测的催化剂活性的倾向。
[0212]
另外,通过h2脉冲化学吸附法求出的co分散度的值(d
ads
),与根据通过tem像求出的co粒子的平均粒子径所预测的co分散度的值(d
tem
)之比,优选满足0<d
ads
/d
tem
<1。co分散度,用露出于金属担载物表面的co的原子数,与金属担载物中包含的全部的co原子数之比表示。co分散度,能够根据担载有co的金属担载物的氢吸附量求出。
[0213]
具体地,假设1个co原子吸附1个h原子,则露出于金属担载物表面的co原子个数所相当的氢原子个数h,与金属担载物担载的co的全部原子个数co之比(h/co),是co分散度。在本发明中,将该基于氢吸附量的co分散度记做d
ads
。通过比较担载相同量(相同原子个数)的co的金属担载物,能够发现,co分散度越高则催化剂活性点数越多。
[0214]
另外已知,若假设co粒子的形态为立方体,则使用通过tem观察求出的co的平均粒子径(d,单位为nm)能够几何学地求出co分散度的值(参照文献《催化剂的辞典》)。该计算方法,能够通过通式(8)表示。co的平均粒子直径,能够从tem像中随机地抽取100~150点的co粒子,并测定各个粒子直径后计算它们的平均值,由此算出。在本发明中,基于通式(4)求出的co分散度的值记做d
tem
。
[0215]dtem
=0.732/d
ꢀꢀꢀ
(8)
[0216]
因此,d
ads
/d
tem
低于1意味着,co粒子的一部分,主要是粒子与复合氧化物(载体)的界面附近、和粒子表面被金属元素b的氧化物覆盖,而阻碍h原子向co粒子表面的吸附。
[0217]
tof(催化剂旋转频率)表示,在1个催化剂表面的活性点上在单位时间内进行的反应的次数。在本技术中,记为在1个作为活性点的表面co的原子上在1秒钟内生成的氨分子的个数。
[0218]
在复合氧化物上担载的co的平均粒子直径,优选为100nm以下,更优选为50nm,还更优选为20nm以下。co的粒子直径越小则在作为氨合成催化剂使用的情况下活性点个数越多,因此是有利的。co的平均粒子直径的下限,没有特别限制,例如是0.5nm以上,也可以是1nm以上。
[0219]
本发明的金属担载物,是担载的金属钴的平均粒子直径为100nm以下的微粒子。由此,在温和的氨合成条件(300~500℃、0.1~20mpa)下,示出了非常高的氨合成速度。
[0220]
需要说明的是,在本说明书中,为了使得表述简洁,将用“担载有co的ba
0.01
mg
0.99o1.00”表示的金属担载物记做“co/ba
0.01
mg
0.99o1”,并将该金属担载物被还原处理后的产物记做“co/ba
0.01
mg
0.99ox”。其他的担载物也使用同样的表示方法。这里x意味着,伴随着还原,烧制时的氧的摩尔比1.00降低为x。需要说明的时候,在本说明书中,在仅仅记做abo
x
的情况下,意味着a与b的量比没有特别限定,并不是a
1.00b1.00ox
的意思。
[0221]
表示复合氧化物中的氧o的比例的通式(2)中的x,表示复合氧化物保持电中性所必须的氧原子的个数。虽然取决于a、b元素的种类,但是x通常在0.5<x≤2的范围内,特别地在0.9<x≤1的范围内。
[0222]
<还原温度对氨合成活性的影响>
[0223]
本发明的催化剂,通过在高温下进行氢还原前处理从而被活性化。这是由于,co被
还原。另外,此时,发现了金属元素a的氧化物堆积于co粒子的表面的特征性的结构。通常若还原前处理为高温,则伴随载体的烧结,会发生比表面积减少、以及金属粒子直径的肥大化,并会引起催化剂活性的降低。
[0224]
图4和图5是示出,在下文所述的实施例(co/bamgo
x
)中,改变还原温度而制造的催化剂的氨合成活性的图表。根据该图表可知,伴随着还原温度的上升而氨合成活性上升,在700℃以及800℃下还原时氨合成速度变大,在900℃下进行还原的情况下,氨生成速度与800℃相比略微降低。
[0225]
图6,示出了下文所述的实施例中的co/bamgo
x
的xrd图谱。根据该图可知,在还原前,可观测到来自于baco3的峰。若还原温度高至700℃以上,那么baco3的峰会消失,因此可知baco3被分解了。另外可知,若升高还原温度,则co的峰变高。
[0226]
另外,如图12所示,可知,在500℃下进行了72小时还原的催化剂,示出了与在700℃下进行1小时的还原的催化剂相匹敌的性能。由此可知,本发明的催化剂,如果是低温还原则能够以长时间获得高活性,如果是高温还原则能够以短时间获得高活性。
[0227]
当还原温度升高时,有tof(催化剂旋转频率)升高的倾向。
[0228]
即,可以认为,当还原温度升高至大于800℃时氨生成速度降低的理由是:载体的粒子的烧结肥大化导致比表面积降低且co的烧结不断进展,并且co粒子的表面过多地被金属元素a的氧化物覆盖,活性点数減少。
[0229]
通过将担载有co的金属担载物用作催化剂,能够使氮与氢反应以制造氨。氨的合成方法自身没有特别限定,例如,通过向装填有催化剂的反应容器内,供应由氢气和氮气构成的原料气体,能够制造氨。反应温度,优选为300~550℃,更优选为300~500℃,还更优选为300~450℃。
[0230]
使用本发明的催化剂进行氨合成时的反应压力,优选为低压的0.1~20mpa,更优选为0.1~15mpa,还更优选为0.1~10mpa。如图10和图11所示,可知担载co的催化剂,与担载ru的催化剂相比,在高压的反应条件下活性也升高。这是由于,与ru相比co难以受到氢中毒的影响,因此在高压下活性也不容易降低。即,在ru的情况下,在高压下表面上吸附的氢原子与ru的相互作用变强,氢原子的脱离非常难产生,ru表面的活性点被闭塞,因氮分子被吸附于该活性点而开始的氨合成反应难以发生。与ru相比,co与氢的相互作用较弱,因此难以产生这样的现象,具有在高的压力下氨合成活性也不容易降低的性质。氨合成反应,通常根据热力学平衡,存在高压越高则氨收获率越增大的倾向,因此通过使用本发明的氨合成用催化剂,可期待在例如10mpa左右的高压条件下得到进一步更高的收获率。另外,与ru相比,co的地壳存在度为其10000倍以上而大量地存在,因此与ru相比具有更高的通用性且成本更低。
[0231]
在使用担载有co的金属担载物作为催化剂的情况下,基于催化剂活性的观点,成为载体的复合氧化物中含有ba是优选的。基于该组合,使用比ru更便宜的co,可发挥充分的氨合成活性。另外,即使在反应压力高时,也不像ru催化剂那样容易受到氢中毒。因此,反应压力最优选为1~10mpa。
[0232]
<复合氧化物
·
金属担载物的制造方法>
[0233]
接着,说明本发明的复合氧化物以及金属担载物的制造方法。本发明的复合氧化物,能够通过如下的方法进行制造。
[0234]
(a)浸渍步骤,使包含金属元素l的l前体含浸包含所述金属元素n的n前体;
[0235]
(b)复合氧化物烧制步骤,在500℃以上的温度下对该混合物进行烧制得到由复合氧化物构成的载体;
[0236]
本发明的金属担载物,进一步能够对于通过上述(a)以及(b)得到的复合氧化物,使用如下的方法,由此进行制造。包括:
[0237]
(c)担载步骤,让所述复合氧化物含浸包含所述金属粒子m的化合物的前体得到含浸载体;
[0238]
(d)担载物烧制步骤,在400℃以上的温度下对所述含浸载体进行烧制。
[0239]
以下,对步骤(a)进行说明。步骤(a)是本发明的复合氧化物的制造方法。复合氧化物,是对包含金属元素l的l前体和包含金属元素n的n前体进行混合并含浸,而得到混合物(复合氧化物的前体)的步骤。
[0240]
复合氧化物的前体,能够通过沉淀法、络合物聚合法等各种方法进行配制。例如,能够使用将氨、氢氧化钠、氢氧化铯等的沉淀剂,与a、b的硝酸盐、氯化物、醋酸盐、碳酸盐、硫酸盐进行反应得到氢氧化物的中和沉淀法。
[0241]
通过单独配制包含金属元素l、金属元素n的元素中的1种以上的产品,并将其进行混合,也能够得到复合氧化物的前体。如此,将包含金属元素l的化合物与包含金属元素n的化合物进行混合,得到混合物。
[0242]
接着,对步骤(b)进行说明。本步骤,是对步骤(a)中得到的混合物进行烧制的步骤。由此,将生成的混合物(复合氧化物前体),通过烧制变为高比表面积的复合氧化物。
[0243]
烧制,在200~400℃左右的低温下优选进行约1~10小时,在400~600℃左右的中间温度下优选进行约1~10小时,在600~700℃左右的高温下优选进行约1~10小时。最终步骤的烧制温度,最优选为700℃。该烧制,只要在空气中、惰性气体与氧的混合气体等含有氧的气氛下进行即可,能够在任意的氧浓度下进行。
[0244]
以下,对步骤(c)进行说明。在步骤(c)中,将步骤(b)中得到的复合氧化物,与溶解有钴、铁、镍等过渡金属粒子供应源的溶剂一起搅拌,从而使得过渡金属粒子供应源含浸于复合氧化物,之后通过进行加热除去溶剂并在该除去后进行过渡金属粒子供应源的分解,从而可得到在复合氧化物载体上以微细的粒子状担载过渡金属粒子的还原处理前担载物。
[0245]
作为钴的过渡金属粒子供应源(钴供应源),能够列举含有co的各种化合物,例如,能够列举乙酰丙酮钴(ii)等有机金属化合物。其中,基于氨合成活性的高度的点,特别优选酰丙酮钴(ii)。还能够使用能够在复合氧化物上担载钴的除此之外的钴供应源,例如硝酸钴、氯化钴、亚硝酰基硝酸钴等。
[0246]
在使用乙酰丙酮钴(ii)之类的有机金属化合物作为钴供应源的情况下,使用有机溶剂作为溶剂是有利的。作为有机溶剂的例子,能够列举四氢呋喃(thf)、甲醇、乙醇、己烷、甲苯等。这些溶剂,如果是一般的的市售品,虽然无需特别地进行前处理就能够使用,但是更优选进行精制、脱水等后使用。与1升溶剂相对的复合氧化物和钴供应源的固体成分浓度,通常优选分别为1~30g/升和0.1~3g/升左右,更优选分别为10~30g/升和0.1~0.3g/升左右。搅拌,能够在常温下进行,搅拌时间优选为1~24小时,更优选为6~12小时。溶剂的除去,能够通过使用各种方法进行加热来进行,优选在例如使用蒸发器等的減压、低温的气氛下进行。钴供应源的分解,通过在惰性气氛例如氦气、氩气或氮气气氛中的加热来进行。
也能够在包含氢的气氛中实施。加热,在200~600℃左右的温度下进行约1~12小时。更优选的加热温度是300~500℃左右,更优选的加热时间是约3~6小时。
[0247]
作为钌供应源,能够使用含有ru的各种各样的化合物。优选地,能够使用十二羰基三钌、乙酰丙酮钌等有机金属化合物。还能够使用能够在复合氧化物上担载钌的除此以外的钌供应源,例如氯化钌、亚硝酰硝酸钌等。
[0248]
以下,对步骤(d)进行说明。接下来,对如此得到的还原处理前担载物(含浸载体)进行还原处理。进行还原处理的目的是,还原过渡金属粒子,以及为了破坏下文所述的碳酸盐。还原温度,为400℃~800℃,优选为600~700℃。在还原温度为超过500℃的高温的情况下,通常还原时间为从10分钟到40小时,优选为30分钟~5小时左右。在还原温度为低温的情况下,还原时间为48小时到120小时,优选为60小时到100小时。还原处理,能够在氢气等还原性气体的存在下进行。
[0249]
已知,在含有强碱性的ba的情况下,bao与空气中的二氧化碳等反应,容易形成碳酸钡(ba(co3))和氢氧化钡(ba(oh)2))。若像这样形成碳酸盐和氢氧化物,则bao的氧的部分负电荷显著降低,无法得到高的碱性。因此,为了显现高的氨合成活性,需要通过适当的处理破坏该碳酸盐和氢氧化物。例如,作为破坏碳酸ba形成bao的方法,在氢气流通下的加热处理(还原处理)是有效的。该反应,用下式表示。
[0250]
baco3 4h2→
bao ch4 2h2o
ꢀꢀꢀ
(5)
[0251]
通过在氢气气氛下加热催化剂,在所担载的金属种的表面会发生氢的解离,会产生还原力强的氢种。在该氢种的作用下,碳酸ba被破坏而变成bao。
[0252]
作为破坏碳酸ba的方法,能够列举在550℃以上的温度下,在氢气流通下将催化剂保温1h左右。优选的条件是600℃到800℃左右。
[0253]
另外,通过在低温下并且在氢气流通下长时间保温催化剂,也能够破坏碳酸ba。优选的条件为500℃下48小时左右,450℃下72小时左右,400℃下120小时以上。
[0254]
通过使用这样的方法,能够破坏ba的碳酸盐。为了显现ba的碱特性,优选尽可能地降低以碳酸盐的形式存在的ba的比例。催化剂中以碳酸盐的形式存在的ba的比例,与催化剂中包含的ba整体的量相比,优选为10mol%以下,更优选为1mol%以下,还更优选为0.1mol%以下,特别优选为0.01mol%以下。
[0255]
作为步骤(d)中的烧制温度,最优选为700~800℃。若该步骤的烧制温度过高,则还原处理时载体以及活性金属会过度烧结,粒子直径变大,故而活性点数減少并且催化剂性能降低。
[0256]
另一方面,若该步骤的烧制温度过高,则载体的比表面积会变小,因此活性金属的分散状态变差、粒子直径变大,故而活性点数減少、催化剂性能降低。
[0257]
对于烧制温度与还原温度的关系,如上文所述,基于氨合成活性的观点,优选在还原处理温度以上的温度下对载体进行烧制。
[0258]
图35对以上情况进行说明。图35,对金属担载物co/bamgo
x
的制造方法不同所导致的差别进行说明。在发明人的先前研究中,公开了一种ba/ru/mgo(参照wo2019/059190的实施例80、81),若将此时的制造方法在本技术中实施,则如下文所述。即,将co担载于ba形成图35(1),进一步担载钡形成图35(2)之后,若在例如700℃下进行高温还原,则生成图35(2)的结构体。即,虽然形成了过渡金属co堆积在作为载体的mgo上、并且氧化钡堆积在过渡金
属co之上的结构,但是根据断面图35(3)可知co与mgo直接接触。
[0259]
另一方面,通过本发明的方法制造金属担载物时,如下所述。即,由于在mgo上担载bao之后担载co所以形成图35(4),之后,将图35(4)例如在700℃下还原,则生成图(5)。这是由于,在高温还原处理中担载的ba化合物获得流动性并移动,从而以覆盖co的方式进行移动。因此,观察断面图35(6)可知,mgo与co没有直接接触,co通过ba化合物而配置于mgo。由于氧化钡以适当的密度覆盖co,因此氮和氢能够到达作为活性点的co上。
[0260]
如此得到的本发明的金属担载物,与目前一直用于氨合成用催化剂的金属担载物相比,操作容易性和反应中的稳定性良好。
[0261]
需要说明的是,在复合氧化物中含有ba等的情况下,即使在制造时催化剂处于氧化状态,如果暴露于大气那么也容易吸收co2而形成碳酸盐。因此,通过上述的还原处理分解碳酸ba后,直到使用催化剂为止,优选以不会暴露在co2中的方式进行处理,优选例如将催化剂密封在填充有惰性气体等的容器中等进行保存。然而,在假设载体的一部分变成碳酸盐的情况下,通过加氢化而进行分解,能够减少碳酸盐并且使得氨合成活性恢复。
[0262]
装填在合成反应器中作为催化剂使用的金属担载物,定期的更换是不可避免的,由于还设想可长时间使用,因此需要操作容易并且稳定性优良的金属担载物。本发明金属担载物,在这一点上是有利的。
[0263]
实施例
[0264]
接着,通过实施例进一步说明本发明。当然,本发明不限于这些实施例。
[0265]
<氨合成活性的测定>
[0266]
使用固定床流通式反应装置,进行金属担载物的氨合成活性的测定。将通过实施例以及比较例记载的方法进行前处理后的金属担载物,一边流通ar一边放置冷却到300℃为止。一边将金属担载物层的温度保持在300℃一边供应ar,同时通过反应管出口的背压阀将压力加压到1.0mpa或3.0mpa为止。停止ar的装入,一边保持压力一边使h2、n2分别以90ml min
-1
、30mlmin
-1
(空间速度72l h
-1g-1
)流通,向反应气氛转变。根据nh3合成活性的高低将1~100mm(1、5、10、25、100mm)的硫酸水溶液200ml加入与导电度计连接的三口烧瓶中,将从反应管出口流出的包含氢(纯度为99.995%,福冈氧制造)、氮(纯度为99.995%,福冈氧制造)、和nh3的混合气体在硫酸水溶液中鼓泡。另外,在除去水分、氧等杂质的情况下使用气体精制器(气体精制过滤器mc50-904f,saes公司制造),使得纯度为99.99999999以上。此时,通过测定nh3与硫酸的反应引起的导电度的变化,对出口气体中包含的氨生成量进行定量。接着,将金属担载物层的温度升温到350℃、400℃或者450℃为止。金属担载物层的温度稳定在350℃、400℃或者450℃后,放置10分钟,使用与上述同样的方法对氨生成量进行定量。
[0267]
<粉末x射线衍射>
[0268]
通过smartlab x-ray diffractometer(理学公司),测定金属担载物(催化剂)的粉末x射线衍射图谱。
[0269]
<比表面积(ssa)测定>
[0270]
金属担载物的比表面积,使用bel-sorp mini(日本bel),基于77k下的氮吸附量通过bet法求出。测定前,作为前处理进行2小时的300℃下的真空加热。
[0271]
1.co/ba
0.01
mg
0.99ox
_700℃还原与co/ba
0.05
la
0.95ox
_700℃还原的比较
[0272]
(实施例1)
[0273]
<co/ba
0.01
mg
0.95ox
_700℃还原>
[0274]
<复合氧化物的配制>
[0275]
ba
0.01
mg
0.99ox
复合氧化物如下合成。将ba(oh)2(和光纯药工业)溶解在净化水中,得到ba(oh)2水溶液,配制200ml的含有0.000625mol的ba的前体溶液。在其中添加2.5g的mgo(宇部兴产),使用磁性搅拌器以320rpm进行搅拌,在室温下持续搅拌1小时。将悬浊液使用旋转蒸发器进行蒸发干固后,使用设定为80℃的炉子将得到的粉体干燥1晩。用研钵将干燥后的沉淀物粉碎,使用电炉将得到的粉体在700℃下在大气氛围下加热5小时,由此得到ba
0.01
mg
0.99ox
。
[0276]
<co的担载>
[0277]
通过含浸法,进行co在载体ba
0.01
mg
0.99ox
上的担载。在200ml茄形瓶中配制溶解有作为co前体的乙酰丙酮钴(ii)(和光纯药工业)的四氢呋喃(thf)(和光纯药工业)溶液,在其中添加1g载体,并在室温下进行18小时以上的搅拌。需要说明的是,适当调节所使用的乙酰丙酮钴(ii)和载体的量,以使得在下述的氩气气氛下加热后的催化剂中包含的co的量为20重量%。将搅拌后的悬浊液使用旋转蒸发器在35℃、0.3atm的条件下进行減压干固后,使用炉子在80℃下干燥18小时。将得到的粉体使用管状电炉在80ml min
-1
的氩气流通下以500℃加热5小时,从而除去前体中的乙酰丙酮配体。通过以上的操作,得到co/ba
0.01
mg
0.99ox
金属担载物。
[0278]
<氢还原前处理>
[0279]
对于上述得到的co/ba
0.01
mg
0.99ox
,使用以下的方法进行氢还原前处理(简称为“前处理”。)。将金属担载物的粉体在20mpa下加压5分钟制作盘体后,用研钵将该盘体粉碎,并使用筛子进行分级制作小丸。小丸的大小调节成以直径计为250~500μm。使用100mg小丸,填充在直径7mm的inconel(
インコネル
(商标))制造的催化剂反应管中,并用石英棉固定催化剂层的前后。将该反应管设置在氨合成活性测定用的固定床流通式反应装置中,在填充有小丸的反应管中流通60ml min
-1
的h2,在700℃下加热1小时,得到co/ba
0.01
mg
0.99ox
_700℃还原。
[0280]
(比较例1)
[0281]
<co/ba
0.05
la
0.95ox
_700℃还原>
[0282]
将专利文献6(国际公开第2019/216304号)的实施例101的co/ba
0.05
la
0.95ox
_700℃还原当做比较例。通过本文献中记载的方法,制造co/ba
0.05
la
0.95ox
_700℃还原。
[0283]
关于实施例1和比较例1的金属担载物,在反应压力1mpa和各种温度(300℃、350℃、400℃、450℃)下进行氨合成,通过上述的方法测定氨合成活性。其结果在下表中示出。
[0284]
【表3】
[0285][0286]
另外,其结果在图1中示出。该图的纵轴右侧表示氨合成速度,纵轴左侧表示氨的收获率,横轴表示反应温度。根据该图可知,在任何的反应温度下,实施例1的金属担载物在氨合成速度和收获率方面,超过了比较例1的金属担载物。还可知,氨合成的反应温度越高,氨合成活性(合成速度,收获率)越高。
[0287]
2.ba添加量的讨论(实施例1~6、比较例2)
[0288]
在实施例1中,对与ba和mg的合计摩尔数相比的ba的摩尔数的比例进行各种改变(0mol%(比较例2)、0.5mol%(实施例2)、1mol%(实施例1)、2mol%(实施例3)、3mol%(实施例4)、5mol%(实施例5)、10mol%(实施例6))从而制造金属担载物。
[0289]
关于实施例1~6和比较例2的金属担载物,在反应压力1mpa和各种温度(300℃、350℃、400℃、450℃)下进行氨合成,通过上述的方法测定氨合成活性。其结果在下表中示出。
[0290]
【表4】
[0291][0292]
另外,其结果在图2和图3中示出。基于该图可知,虽然随着ba的添加量的增多,氨合成活性升高,但是在添加量为1mol%时氨合成活性最大,当添加量增加到超过该量时,氨合成活性会逐渐降低。另外,根据图3可知,氨合成活性高的ba的添加量,在0.5~3mol%的范围内。
[0293]
3.还原温度的讨论(实施例7~11)
[0294]
在实施例1中,将co的担载量设为20wt%,对还原温度进行各种改变(500℃(实施例7)、650℃(实施例8)、700℃(实施例9)、800℃(实施例10)、900℃(实施例11))从而制造金属担载物。
[0295]
关于实施例7~11的金属担载物,在反应压力1mpa和各种温度(300℃、350℃、400℃、450℃)下进行氨合成,通过上述的方法测定氨合成活性。其结果在下表中示出。
[0296]
【表5】
[0297][0298]
另外,其结果在图4和图5中示出。根据该图可知,虽然还原温度为500℃时,氨合成活性比较低,但是在650℃以上时氨合成活性升高。
[0299]
图6示出了实施例7(还原温度500℃)、实施例9(还原温度700℃)、实施例10(还原温度800℃)中的金属担载物的xrd图谱。图中的“fresh”是指还原处理前的样品。如根据该图可知,还原处理导致baco3分解,当将还原温度升高到700℃左右时,未能观测到baco3的峰,因此可知baco3被分解。另外可知,当将还原温度升高到800℃时,可观测到目前为止未能观测的金属co的峰。可认为其原因是,由于co粒子高度地分散所以未能观测的金属co的峰,在高温还原处理的作用下凝集成略微分散的co粒子时可观测。
[0300]
下表,示出了实施例7(还原温度500℃)、实施例9(还原温度700℃)、实施例10(还
原温度800℃)、比较例2(无ba,还原温度700℃)在反应温度350℃下的金属担载物的各种参数。根据该表可知,虽然随着还原温度的升高而比表面积(ssa)降低,但是氨合成活性升高。
[0301]
【表6】
[0302]
co/bamgox催化剂还原温度讨论各还原温度参数(350℃)
[0303][0304]
4.co担载量的讨论(实施例12、实施例1、实施例15)
[0305]
在实施例1中,对co的担载量进行各种改变(5wt%(实施例12)、10wt%(实施例13)、20wt%(实施例1)、30wt%(实施例15))从而制造金属担载物。
[0306]
关于实施例12、实施例1、实施例15的金属担载物,在反应压力1mpa和各种温度(300℃、350℃、400℃、450℃)下进行氨合成,通过上述的方法测定氨合成活性。其结果在下表中示出。
[0307]
【表7】
[0308][0309]
另外,其结果在图7和图8中示出。根据该图可知,随着co担载量增多而氨合成活性升高,在20wt%下氨合成活性大致为最大。
[0310]
(比较例1、4、5)
[0311]
作为比较例,制作co/ba
0.05
la
0.95ox
(比较例1)、ru/ceo
x
、(比较例4)、cs
/ru/mgo
x
(比较例5)。
[0312]
ru/ceo
x
、(比较例4),通过以下的方法制成。通过含浸法,进行ru担载于ceo2。在200ml茄形瓶中配制溶解有作为ru前体的ru3(co)
12
(古屋金属)的四氢呋喃(thf)(和光纯药工业)溶液,在其中添加5g的ceo2(第一稀元素化学工业),在室温下搅拌18小时以上。需要说明的是,适当调节所使用的ru3(co)
12
和载体的量,以使得在氩气气氛下加热后的催化剂中包含的ru的量为5重量%。将搅拌后的悬浊液使用旋转蒸发器在35℃、0.3atm的条件下进行減压干固后,使用炉子在80℃下干燥18小时。将得到的粉体,使用管状电炉在500℃、80mlmin
-1
的氩气流通下加热5小时,由此除去前体中的羰基配体。通过以上的操作,得到ru/ceo2。还原处理在400℃下进行。
[0313]
cs
/ru/mgo
x
(比较例5),通过非专利文献(f.rosowski,a.hornung,o.hinrichsen,d.herein,m.muhler and g.ertl,appl.catal.,a,1997,151,443-460.)中记载的方法制作。需要说明的是,ru的担载量设为5重量%,cs/ru设为1/1(mol/mol)。还原处理在500℃下进行。
[0314]
关于比较例3~5的金属担载物,在各种温度(300℃、350℃、400℃、450℃)和压力(0.1mpa、1.0mpa、3.0mpa)下进行氨合成,通过上述的方法测定氨合成活性。其结果在下表
中示出。
[0315]
【表8】
[0316][0317]
另外,其结果在图10(反应温度350℃)以及图11(反应温度400℃)中示出。实施例3(co/ba
0.01
mg
0.99ox
)的结果也一并在图中示出。根据该图可知,与比较例1、4、5的金属担载物相比,实施例的金属担载物的氨合成活性更高。
[0318]
6.前处理条件(还原条件)的影响(实施例1、实施例17、比较例6)
[0319]
在实施例1中,将氢还原前处理的温度和时间改变为500℃、72小时,制作金属担载物(实施例17)。作为比较例,5wt%ru/mgo(比较例6),除了选用mgo(宇部材料)作为载体以外,使用与比较例4同样的方法进行制作。
[0320]
关于实施例1和实施例17、比较例6的金属担载物,在反应压力1mpa和各种温度(300℃、350℃、400℃、450℃)下进行氨合成,通过上述的方法测定氨合成活性。其结果在下表中示出。
[0321]
【表9】
[0322][0323]
另外,其结果在图12中示出。根据该图可知,即使还原条件为低压,通过长时间的还原氨合成活性也会提高。
[0324]
7.co前体的影响(实施例1、实施例18)
[0325]
在实施例1中,除了使用co(no3)2·
6h2o(和光纯药)代替作为co前体的co乙酰丙酮(ii)(co(acac)),使用净化水代替thf,并且烧制时的气氛选用空气以外,使用与实施例1相同的方法,制作金属担载物(实施例18)。
[0326]
关于实施例1和实施例18的金属担载物,在反应压力1mpa和各种温度(300℃、350℃、400℃、450℃)下进行氨合成,通过上述的方法测定氨合成活性。其结果在下表中示出。
[0327]
【表10】
[0328][0329]
另外,其结果在图13中示出。根据该图可知,关于co前体,与硝酸co相比co(acac)更能提高氨合成活性。
[0330]
图14示出实施例1(co(acac))、实施例17(硝酸co)的金属担载物的xrd。根据该图可知,在选用co(acac)作为前体的金属担载物中,未能观测来自金属co的峰,但是在选用硝酸作为前体的金属担载物中,观测到来自金属co的峰。由此可推测,与选用硝酸co作为前体的金属担载物相比,选用co(acac)作为前体的金属担载物,co分散更高,其结果是氨合成活
性升高。
[0331]
8.sv讨论(实施例1)
[0332]
在实施例1中,代替作为co前体的co乙酰丙酮(ii)(co(acac))而使用co(no3)2·
6h2o(和光纯药),制作金属担载物(实施例18)。
[0333]
关于实施例1的金属担载物,在反应压力1mpa和各种温度(300℃、350℃、400℃、450℃)和各种sv(18l/h
-1g-1
,36l/h
-1g-1
,72l/h
-1g-1
)下进行氨合成,通过上述的方法测定氨合成活性。其结果在下表中示出。
[0334]
【表11】
[0335][0336]
另外,其结果在图15和图16中示出。根据该图可知,虽然随着sv的值增大氨合成速度增加(图15),但是氨收获率降低(图16)。
[0337]
9.载体特性的讨论(实施例1、比较例7、比较例1)
[0338]
(5wt%ru/ba
0.1
la
0.45
ce
0.45ox
:比较例7)
[0339]
在比较例1中,代替ceo2使用ba
0.1
la
0.45
ce
0.45
,并适当调节ru含量以制作5wt%ru/ba
0.1
la
0.45
ce
0.45ox
(比较例7)。需要说明的是,载体,根据专利文献6(国际公开第2019/216304号)的实施例6中记载的方法,使用反均匀沉淀法如下合成。将la(no3)3·
6h2o(和光纯药工业)溶解在净化水中,形成la(no3)3水溶液。将ce(no3)3·
6h2o(关东化学)溶解在净化水中,形成ce(no3)3水溶液。将ba(no3)2·
6h2o(和光纯药工业)溶解在净化水中,形成ba(no3)2水溶液。混合la(no3)3水溶液和ce(no3)3水溶液,并混合ba(no3)2水溶液,配制250ml的合计含有0.0625mol的la和ce和ba的载体前体溶液。将28%nh3水溶液(和光纯药工业)在1000ml烧杯中添加250ml,一边使用磁性搅拌器以320rpm进行搅拌,一边一次性地添加上述载体前体溶液,进行1小时的搅拌。之后静置12小时,通过吸引过滤分离沉淀物(1)。将分离后的滤液回收在2l烧杯中。在分离后的沉淀物(1)中添加350ml的离子交换水,搅拌30分钟并清洗沉淀物,通过吸引过滤分离沉淀物(1)。将该清洗的操作进行3次。将清洗中使用的离子交换水全部回收,将滤液和清洗液添加到2l烧杯中并混合后,将该混合溶液放置12小时,使得白色的沉淀物(2)产生,并通过吸引过滤回收产生的沉淀物(2)。将沉淀物(1)与沉淀物(2)混合,使用炉子在80℃下干燥15小时。用研钵将干燥后的沉淀物粉碎,使用电炉将得到
的粉体在700℃下在大气气氛下加热5小时,由此得到ba
0.1
la
0.45
ce
0.45ox
。
[0340]
关于实施例1、比较例1、比较例7的金属担载物,在反应压力1mpa、反应温度350℃下进行氨合成,使用上述的方法测定氨合成活性。其结果在下表中示出。
[0341]
【表12】
[0342]
co/bamgox催化剂还原温度讨论(700℃,1h,red.)(1.0mpa)
[0343][0344]
根据该表,实施例1的20wt%co/ba
0.01
mg
0.99ox
,比表面积(ssa)较大,可推测这有助于氨合成活性的升高。
[0345]
10.低温下的活性(实施例1、比较例9、比较例10)
[0346]
作为比较例,使用专利文献6(国际公开第2019/216304号)的实施例6中记载的方法,制造ru/ba
0.1
la
0.45
ce
0.45ox
(700℃、1h、red)(比较例9)。
[0347]
另外,使用专利文献6(国际公开第2019/216304号)的实施例1中记载的方法,制造ru/la
0.5
ce
0.5ox
(650℃、1h、red)(比较例10)。
[0348]
关于实施例1,在各种温度(150℃、200℃、250℃)和各种压力(0.1mpa、1.0mpa、3.0mpa)下进行氨合成,通过上述的方法测定氨合成活性。另外,关于比较例9、比较例10的金属担载物,在各种温度(150℃、200℃、250℃)下进行氨合成(压力1.0mpa),利用上述的方法测定氨合成活性。其结果在下表中示出。
[0349]
【表13】
[0350][0351]
另外,其结果在图17、图18中示出。根据图17可知,在低温条件下,co担载催化剂示出了比ru担载催化剂更高的活性。另外,根据图18可知,在低温条件下,随着压力的增加氨合成活性升高。
[0352]
11.ru担载催化剂的讨论(实施例21、比较例11~比较例14)
[0353]
(实施例21)
[0354]
<ru/ba
0.01
mg
0.99ox
_700℃还原>
[0355]
除了在实施例1中,将乙酰丙酮co(ii)替换为作为ru前体的ru3(co)
12
(古屋金属)以外,进行与实施例1相同的操作,得到ru/ba
0.05
mg
0.95ox
_700℃还原。
[0356]
11.fe担载催化剂的讨论(实施例22、比较例15、比较例16)
[0357]
(实施例22)
[0358]
<fe/ba
0.01
mg
0.99ox
_700℃还原>
[0359]
除了在实施例1中,将乙酰丙酮co(ii)替换为作为fe前体的乙酰丙酮铁(iii)(同仁化学研究所)以外,进行与实施例1相同的操作,得到fe/ba
0.01
mg
0.99ox
_700℃还原。
[0360]
(比较例15、比较例16)
[0361]
作为比较例,除了将ru替换成fe,并且改变ba的量以外,按照与比较例9相同的方法,制作20wt%fe/ba
0.1
la
0.45
ceo
x
(700℃,1小时还原)(比较例15)。另外,通过与比较例6相同的方法,制作5wt%ru/mgo(比较例16)。
[0362]
关于实施例22、比较例15、比较例16的金属担载物,在反应压力1mpa和各种温度(300℃、350℃、400℃、450℃)下进行氨合成,通过上述的方法测定氨合成活性。其结果在下表中示出。
[0363]
【表14】
[0364][0365]
另外,其结果在图20中示出。根据该图可知,担载有fe的金属担载物,与5wt%ru/mgo和20wt%fe/ba
0.1
la
0.45
ceo
x
(700℃,1小时还原)相比,氨合成活性更高。
[0366]
12.前处理条件的讨论:fe担载催化剂(实施例23、实施例24、比较例16)
[0367]
在实施例22中,对前处理的条件进行各种变更(h2 n2、500℃、72小时还原(实施例23);仅有h2、700℃、1小时还原(实施例24)),制造金属担载物。
[0368]
关于实施例23、实施例24、比较例16的金属担载物,在反应压力1mpa和各种温度(300℃、350℃、400℃、450℃)下进行氨合成,通过上述的方法测定氨合成活性。其结果在下表中示出。
[0369]
【表15】
[0370][0371]
另外,其结果在图21中示出。根据该图可知,取决于前处理条件,而氨合成活性大幅提高。
[0372]
13.co-fe催化剂的讨论(实施例25~实施例27)
[0373]
在实施例7(20wt%co/ba
0.05
mg
0.95ox
)中,在co前体中添加实施例22的fe前体对混合比进行配制,制造金属担载物(实施例25)。
[0374]
关于实施例7、实施例22、实施例25的金属担载物,在各种温度(300℃、350℃、400℃、450℃)下进行氨合成,通过上述的方法测定氨合成活性。其结果在下表中示出。
[0375]
【表16】
[0376][0377]
另外,其结果在图22中示出。根据该图可知,与单独fe和fe-co混合催化剂相比,单独co的氨合成活性更高。
[0378]
14.h2-tpr测定结果(实施例1、比较例2)
[0379]
对于实施例1(co/ba
0.05
mg
0.95ox
_700℃还原)、比较例2(co/mgo
x
),进行h2-tpr(h-temperature programmed reduction:h-程序升温还原)测定。h2-tpr是指,在使用氩气等惰性气体稀释后的氢气(h2)的流通下,使得固体的温度以一定速度连续地上升,对氢气的消耗速度、反应生成物的生成速度进行计测,并且使用质量分析计作为检测器。其结果在图23、图24中示出。
[0380]
根据图23可知,实施例1的co/bamgo
x
,在500多℃下吸收分子量为2的氢,并释放出分子量为16的甲烷。另外可知,在600多℃下释放出分子量为18的水。另一方面,根据图24可知,比较例2的co/mgo
x
,没有这样的吸收释放。由这些可知,在实施例1中,关于ba发生了下式(5)的还原反应。
[0381]
baco3 4h2→
bao ch4 2h2o
ꢀꢀꢀ
(5)
[0382]
该结果表示,虽然co/bamgo
x
催化剂在表面生成baco3、ba(oh)2,但是通过氢气下的热处理,在500多℃下h2与baco3反应而分解成ba(oh)2和ch4,在600多℃下ba(oh)2分解成bao和h2o。即,可以认为,通过进行在氢气下的热处理,因而在表面生成的baco3和ba(oh)2变成bao,所以催化剂活性升高。
[0383]
图25,是实施例1的催化剂的电子显微镜照片。图25(1)是haadf-stem图像。根据图25(2)~图25(5)的元素分布图像,可得知如下内容。图25(1)中发光最白的部分为co,接着发浅灰色的光的部分是存在氧化镁的部分。参见图25(5)可知,氧化钡的粒子,以比co的粒子更小的粒子状下,均匀地分别在co/mgo上。氧化钡的粒子,至多是co粒子的粒径的10%左右。催化剂的制作的顺序是,在氧化镁载体上担载氢氧化钡,然后在氢氧化钡上担载钴,之后氧化钡的粒子在催化剂制造条件下移动至钴粒子上,在钴担载前,钡化合物已经均匀
地分别在氧化镁的表面,因此可认为在氧化镁和钴的界面附近也存在ba。根据图25(5)的重叠的图像可知,以ba覆盖co的周围的方式,存在纳米等级的核(co粒子)/壳(氧化钡)结构。需要说明的是,在该催化剂中,接近氧化钡的表面钴原子会受到强的电性供应,因此示出了较高的氨合成活性。这一事实证明了上文的研究。
[0384]
图34,是实施例17的催化剂的电子显微镜照片。图(1)是haadf-stem图像。图34(2)~(5)是元素分布图像,与图25进行比较,可得知以下内容。在500℃、72小时还原(图34)下,与700℃、1小时还原(图25)相比,还原温度更低,因此虽然可以观察到纳米等级的核(co粒子)/壳(氧化钡)结构,但是氧化钡的粒子直径小,氧化钡向钴粒子上的移动度也略低。但是,与长时间进行还原处理相应地,钡的碳酸盐以及氢氧化物的分解,比500℃、1小时还原(实施例7)的情况更持续地进行。另一方面,由于还原温度低,因此钴粒子直径小,另外,与长时间进行还原处理相应地,钴的还原度,比500℃、小时还原(实施例7)的情况更高,有助于氨合成的金属状态的表面钴原子的个数增多。因此,实施例17的催化剂,示出了与实施例1的催化剂匹敌的氨合成活性。
[0385]
如下文所述的实施例33、34所示,第1族元素 第2族元素的双添加的情况下,可发现实施例17相比氨合成活性进一步提高。这是由于,氧化钡的壳结构中引入了比第2族元素的氧化物更强的碱性元素的第1族元素,接近壳的表面钴原子受到了非常强的电性供应。
[0386]
15.将ba改变为其他的第二族元素(sr、ca)后的催化剂的氨合成活性(实施例26~27)
[0387]
在实施例1中,代替原料的ba(oh)2,使用sr(oh)2、ca(oh)2,制作20wt%co/sr
0.01
mg
0.99ox
(实施例26)和20wt%co/ca
0.01
mg
0.99ox
(实施例27)。关于这些金属担载物,在各种温度(300℃、350℃、400℃、450℃)下进行氨合成,通过上述的方法测定氨合成活性。氨合成条件,反应压力设为1.0mpa,反应气体选用h2/n2=90/30cc/min(总流量为120cc/min),催化剂量选用0.1g(sv=72l h
-1g-1
)。其结果在下表中示出。
[0388]
【表17】
[0389][0390]
另外,其结果在图26中示出。根据该图可知,关于除了ba以外的第2族元素(sr、ca),与仅仅mgo相比活性更高,可确认添加该元素所得到的氨合成活性提高的效果。
[0391]
16.代替第2族元素而添加第1族元素(实施例28~31)
[0392]
在实施例1中,代替原料的ba(oh)2,使用kno3、koh、lino3、lioh,在各种还原温度下制造复合氧化物,分别制作20wt%co/k
0.03
mg
0.97ox
(实施例28)、20wt%co/k
0.03
mg
0.97ox
(实施例29)、20wt%co/li
0.03
mg
0.97ox
(实施例30),20wt%co/li
0.03
mg
0.97ox
(实施例31)。关于这些金属担载物,在各种温度(300℃、350℃、400℃、450℃)下进行氨合成,通过上述的方法测定
氨合成活性。其结果在图27中示出。根据该图可知,使用koh来添加k的系统,虽然看到了一些氨活性提高的效果,然而不是像第2族元素那样显著提高的氨合成活性。
[0393]
17.第1族元素 第2族元素的双添加(实施例32~34)
[0394]
在实施例1中,除了原料的ba(oh)2之外还使用csoh、rboh、koh制造复合氧化物,在700℃、1小时的还原条件下进行前处理,分别制作20wt%co/cs
0.01
ba
0.01
mg
0.98ox
_700℃、1h还原(实施例32),20wt%co/rb
0.01
ba
0.01
mg
0.98ox
_700℃、1h还原(实施例33),20wt%co/k
0.01
ba
0.01
mg
0.98ox
_700℃、1h还原(实施例34)。关于这些金属担载物,在各种温度(300℃、350℃、400℃、450℃)下进行氨合成,通过上述的方法测定氨合成活性。其结果在下表中示出。
[0395]
【表18】
[0396][0397]
另外,其结果在图28中示出。根据该图可知,在添加1mol%的第1族元素,并且将前处理条件固定为700℃、1h的情况下,其中任一者与没有添加第1族元素的情况(仅仅ba)相比,虽然活性略微降低,但是得到了与仅仅ba的情况相同程度的氨合成活性。其中,由于仅仅ba的情况的氨合成活性最高,所以没有发现添加第1族元素(双添加)而得到的氨合成活性的提高。
[0398]
18.第1族元素 第2族元素的双添加(还原条件的讨论1)(实施例35~36)
[0399]
在实施例30中,增加原料的koh的量,改变还原条件,制作20wt%co/k
0.03
ba
0.01
mg
0.96ox
_500℃、72h还原(实施例35),20wt%co/k
0.03
ba
0.01
mg
0.96ox
_700℃、1h还原(实施例36)。关于这些金属担载物,在各种温度(300℃、350℃、400℃、450℃)下进行氨合成,通过上述的方法测定氨合成活性。其结果在下表中示出。
[0400]
【表19】
[0401][0402]
另外,其结果在图29中示出。根据该图可知,在3mol%k 1mol%ba的情况下,即使在低温下进行长时间还原,也没有发现提高氨合成活性的效果。
[0403]
19.第1族元素 第2族元素的双添加(还原条件的讨论2)(实施例37)
[0404]
在实施例30中,不改变原料的koh的量,而改变还原条件,制作20wt%co/k
0.01
ba
0.01
mg
0.98ox
_500℃、72h还原(实施例37)。关于这些金属担载物,在各种温度(300℃、350℃、400℃、450℃)下进行氨合成,通过上述的方法测定氨合成活性。其结果在下表中示出。
[0405]
【表20】
[0406][0407]
另外,其结果在图30中示出。根据该图可知,在1mol%k 1mol%ba的情况下,若在低温下进行长时间还原,则活性提高。需要说明的是,在图中,在反应温度450℃附近出现平衡的影响,可推测任一者均为同等程度的氨合成活性。
[0408]
20.第1族元素 第2族元素的双添加(还原条件的讨论3)(实施例38)
[0409]
在实施例29中,改变还原条件,制作20wt%co/rb
0.01
ba
0.01
mg
0.98ox
_500℃、72h还原(实施例38)。关于这些金属担载物,在各种温度(300℃、350℃、400℃、450℃)下进行氨合成,通过上述的方法测定氨合成活性。其结果在下表中示出。
[0410]
【表21】
[0411][0412]
另外,其结果在图31中示出。根据该图可知,在1mol%rb 1mol%ba的情况下,若在低温下进行长时间还原,则活性提高。需要说明的是,在图中,在反应温度450℃附近出现平衡的影响,可推测任一者均为同等程度的氨合成活性。
[0413]
21.代替co的ni(反应压力的讨论1)(实施例39)
[0414]
在实施例1中,除了将乙酰丙酮co(ii)替换为作为ni前体的乙酰丙酮镍(ii)(岸田化学)以外,进行与实施例1相同的操作,得到ni/ba
0.01
mg
0.99ox
_700℃、1小时还原(实施例35)。关于该金属担载物(实施例39)、和实施例1、实施例22的金属担载物,在1.0mpa和各种温度(300℃、350℃、400℃、450℃)下进行氨合成,通过上述的方法测定氨合成活性。其结果在下表中示出。
[0415]
【表22】
[0416][0417]
另外,其结果在图32中示出。根据该图可知,ni担载催化剂,在450℃下,氨合成活性比fe担载催化剂更高。
[0418]
22.代替co的ni(反应压力的讨论2)(实施例40)
[0419]
对于实施例1、实施例22和实施例40的金属担载物,在3.0mpa的条件下进行氨合成,通过上述的方法测定氨合成活性。其结果在下表中示出。
[0420]
【表23】
[0421][0422]
另外,其结果在图33中示出。根据该图可知,ni担载催化剂和fe担载催化剂,在3mpa下也分别示出了相当高的活性。其中,对于ni担载催化剂,可观察到稍微受到中毒的影响。
[0423]
23.co的还原度
[0424]
为了比较氢还原处理对催化剂中的co的还原状态带来的影响,对co/bamgo
x
的co k吸收端的x射线吸收端近边结构(xanes)光谱进行测定。
[0425]
关于还原处理前的催化剂以及作为比较对象的标准试料,按照以下的方法配置xanes频谱测定用的样品。在大气中将还原处理前的催化剂以及标准试料与氮化硼的粉末进行磨碎混合,将混合后的粉体加压成形为直径10mm的盘状。此时,对各催化剂以及标准试料与氮化硼之比、以及盘的厚度进行适当调节,以使得测定对象中的co的浓度,对于在频谱测定时透过的x射线的吸光度来说,是最合适的。
[0426]
关于还原处理后的催化剂,按照以下的方法配置xanes频谱测定用的样品。将催化剂填充在试料管中并且与固定床流通式的反应装置连接,一边流通氢气,一边在规定的温度下加热1小时,由此进行还原处理。关于还原处理后的催化剂,一边向试料管中供应惰性气体(ar)一边将其放置冷却到室温为止。在该反应装置的试料管上,在气体的入口和出口侧具备旋塞,在放置冷却后闭合该旋塞,从而即使从反应装置中取出,也能够保持催化剂不与大气接触。将使用该试料管还原后的催化剂转移到被惰性气体填充的手套箱中,在手套
箱内将还原后的催化剂与氮化硼的粉末进行磨碎混合之后,将混合后的粉体加压成形为直径10mm的盘状。此时,对各催化剂与氮化硼之比、以及盘的厚度进行适当调节,以使得测定对象中的co的浓度,对于在频谱测定时透过的x射线的吸光度来说,是最合适的。
[0427]
成形后的盘,在手套箱内被三重封入氧隔绝性树脂袋。由此,即使将树脂袋从手套箱取出到大气中,也能够在催化剂不受到氧的再氧化的影响的情况下测定光谱。
[0428]
所制成的测定用的各样品的xanes光谱,使用大型放射光设施(spirng-8)的bl01b1,进行测定。对于在检测器中使用离子室、并使用透过法测定的光谱,使用x射线吸收光谱分析软件(athena,demeter 0.9.26)进行分析。
[0429]
图36中,示出了标准化的催化剂以及标准试料的xanes光谱。将未还原的催化剂与标准试料的光谱形状进行比较发现,含有ba的情况和不含有的情况,未还原的催化剂的xanes光谱的能量位置以及形状,与氧化物(ii)(coo)的非常一致。因此,这表示未还原的催化剂中,co以coo的形式存在。另外,通过进行还原处理,xanes光谱的形状接近co箔的光谱形状。这意味着,通过还原处理,催化剂中的co变化为金属状态。
[0430]
因此,以标准试料的金属co箔以及co氧化物(ii)的光谱为基准,对还原处理后的各催化剂的标准化后的xanes光谱进行线性组合拟合,找出催化剂中含有的金属状态的co的比例(还原度)。其结果是,co/bamgo
x
催化剂:
[0431]
在500℃、1小时还原后的co还原度为71%,
[0432]
在700℃、1小时还原后(实施例1的催化剂)的co还原度为93%。
[0433]
明显可知,随着还原处理温度上升,对氨合成没有活性的co氧化物的比例减少,另一方面,对氨合成有活性的金属状co的比例增多。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
