alk5抑制剂缀合物及其用途
1.1.相关申请的交叉引用
2.本技术要求提交于2020年1月8日的美国临时申请62/958,461号的优先权权益,其内容通过引用整体并入本文。
2.
背景技术:
3.2.1纤维化
4.特发性肺纤维化(ipf)是破坏性的肺部慢性疾病,其特征是肺组织进行性硬化和瘢痕形成(lederer et al.,2018,nejm,378:1811-23;barratt et al.,2018,j clin med 7(8):201)。在美国,每年诊断出大约130,000名患者,5年死亡率为80%。迄今为止,还没有治愈这种疾病的方法,只有减缓其进展的选择(somogyi et al.,2019,eur respir rev,28(153):190021)。ipf始于外在刺激物(如吸烟)对肺泡上皮的反复损伤,然后是持续的成纤维细胞激活,这是纤维化的驱动因子之一。由于持续的肺损伤,纤维化本质上是无法愈合的伤口。
5.肺成纤维细胞分化成肌成纤维细胞是组织纤维化发展的主要步骤(yazdani et al.,2017,adv drug deliv rev 121:101-116;huang et al.,2014,austin j pulm resp,1(1):3-9)。肌成纤维细胞负责纤维发生并且肌成纤维细胞主要发现在纤维化区域中和在该区域中活跃。有ipf中肌成纤维细胞的三种可能来源:1)驻留的肺上皮细胞,其在称为上皮-间质转化(emt)的过程中转化为肌成纤维细胞;2)转化为肌成纤维细胞(fmt)的驻留肺成纤维细胞;和/或3)募集到肺部以驱动纤维化和瘢痕形成的肌成纤维细胞(pardali et al.,2017,int j mol sci,18(10))。这三种途径的结合导致肺驻留肌成纤维细胞的增加,从而驱动纤维化疾病。抑制肌成纤维细胞将是逆转纤维化肺疾病的重要步骤。
6.多效(pleiotropic)细胞因子转化生长因子-β(tgf-β)负责体内大多数组织的发育、维持和稳态。tgf-β经由与tgf-β受体ii和tgf-β受体i/alk5结合来启动信号传导。alk5是丝氨酸苏氨酸激酶受体,其磷酸化下游信号传导介质smad2和smad3。活化的smad2/3与smad4形成复合物并转移到细胞核中以调节基因表达,这由细胞环境决定(derynck et al.,2003,nature,425(6958):577-84)。在肺中,tgf-β由极其多种细胞类型产生,其包括肺泡巨噬细胞、嗜中性粒细胞、活化的肺泡上皮细胞、内皮细胞、成纤维细胞和肌成纤维细胞(caja et al.,2018,int j mol sci,19(5))。tgf-β是细胞外基质(ecm)产生的最有效诱导剂之一,其包括α-平滑肌肌动蛋白(αsma)、胶原蛋白和纤连蛋白(pohlers et al.,2009,biochim biophys acta,1792(8):746-56;kim et al.,2018,cold spring harb perspect biol,10(4))。在ipf疾病进展期间,tgf-β增加胶原蛋白表达和ecm沉积、肌成纤维细胞扩增、成纤维细胞向肌成纤维细胞转化和上皮-间质转化(emt)(pardali et al.,2017,int j mol sci,18(10);yue et al.,2010,curr enzym inhib,6(2))。此外,tgf-β在肺纤维化动物模型和纤维化人肺中的表达均升高(tashiro et al.,2017,front med(lausanne),4:118)。在肺纤维化动物模型中,tgf-β水平升高先于胶原蛋白合成和沉积。作为tgf-β作为体内肺纤维化驱动因子作用的进一步证据,腺病毒在肺中表达tgf-β1或tgf-β1的转基因肺特
异性表达的动物模型足以驱动肺纤维化(lee et al.,korean j intern med,29:281)。在博莱霉素(bleomycin)诱导ipf的经典小鼠模型中,肺中tgf-β水平升高,通过smad 3敲除小鼠或tgfbrii在成纤维细胞中特异性显性负表达阻断tgf-β信号传导导致疾病严重程度降低(fernandez et al.,2012,proc am thorac soc,9(3):111-116;degryse et al.,2011,am j physiol lung cell mol physiol,300(6):887-897;li et al.,2011,j clin invest,121(1):277-87)。在治疗上,用小分子tgf-β受体抑制剂或抗tgf-β抗体治疗也在博莱霉素和辐射诱导的纤维化中抑制疾病(giri et al.,1993,thorax,48:959-66;flechsig et al.,2012,clin cancer res,18(13):3616-27)。
7.由于tgf-β在驱动ipf中的突出作用,已研究针对tgf-β途径的疗法用于治疗ipf。然而,由于tgf-β及其受体在体内的广泛表达,宿主组织毒性的风险使得开发安全有效的疗法变得困难(anderton et al.,2011,toxicologic path,39:916-24;stauber et al.,2014,clinical tox,4(3):1-10;lonning et al,2011,curr pharma biotech,12:2176-89)。例如,系统性阻断tgf-β活性的αvβ6整合素抗体的2期试验(bg00011)最近因安全问题而终止(arefayene,et al.,2018,european respiratory journal,52(suppl 62)pa596)。像大多数药物一样,毒性和治疗窗口必须平衡;对于广泛作用的tgf-β抑制剂,安全性和毒性风险是最重要的。能安全逆转纤维化的选择性强效tgf-β抑制剂可能会阻止疾病进展,也可能提高患者的存活率。
8.2014年,两种ipf药物获批,吡非尼酮(pirfenidone)(抗纤维化分子),以及尼达尼布(nintedanib)(酪氨酸激酶抑制剂),这两种药物都可能部分阻断tgf-β信号传导以及其他途径(gan et al.,2011,ther clin risk manag,7:39-47;margaritopoulos et al.,2016,core evid,11:11-22;lunardi et al.,2018,arch pathol lab med,142:1090-1097)。一般而言,吡非尼酮和尼达尼布治疗可将轻中度疾病患者的ipf疾病进展的风险减少50%(ren et al.,2017,saudi med j,38(9):889-894;case et al.,2017,bmj open resp res,4:e000192)。然而,将肺功能严重下降《50%的ipf患者(通过fvc、用力肺活量、患者可呼出的空气总量来衡量)、患有并存病的老年患者或未正式诊断为ipf的患者排除在这些试验之外。虽然这两种药物都可以减缓疾病,但它们无法完全阻止或逆转疾病进展。由于纤维化患者仍有大量未满足的需求,ipf的试验引起了极大的兴趣。
9.在治疗ipf方面,证明了其他疗法,如ifn-γ抑制剂、血管生成抑制剂和tnf-α阻断剂是不成功的(yazdani et al.,2017,adv drug deliv rev,121:101-116;somogyi et al.,2019,eur respir rev,28(153):190021)。正在进行的ipf试验包括血清淀粉样蛋白p(pentraxin;ptx-2),它是循环蛋白,可与单核细胞结合并抑制其分化为促纤维化纤维细胞,从而促进上皮愈合和纤维化消退。ipf患者的pentraxin水平较低,正在进行的2期试验表明肺功能和6分钟步行试验得到改善。在一项2期试验中,pamrevlumab是完全重组的针对结缔组织生长因子(ctgf)的人单克隆抗体,其可减少纤维化、减少ipf患者肺功能(fvc)的降低(somogyi et al.,2019,eur respir rev,28(153):190021)。相比之下,降低tgf-β和ccl2表达的il-13抗体tralokinumab和降低ecm交联的抗loxl2抗体simtuzumab均因呼吸功能缺乏改善而在2期试验失败(raghu,2017,european respiratory review,26:170071)。许多疗法直接或间接改变tgf-β功能。然而,尽管做出了这些努力,但对ipf患者的改善治疗的需求仍未得到满足,特别是对于可以选择性和安全地改变疾病的疗法。
10.在ipf以外的多种疾病中,纤维化由tgf-β驱动,其包括其他类型的肺纤维化(例如,与系统性硬化症相关)、肝纤维化(例如,与非酒精性脂肪性肝炎(nash)相关)、肾纤维化和心脏纤维化(meng et al.,2016,nat rev nephrol.12(6):325-38;biernacka et al.,2011,growth factors,29(5):196
–
202;gyorfi et al.,2017,matrix biology,68-69:8-27)。因此,对于能够逆转tgf-β驱动的肌成纤维细胞活化和减少有需要的受试者,特别是患有肺纤维化(例如ipf)、肝纤维化(例如与nash相关的)、肾纤维化、心脏纤维化和系统性硬化症的患者的纤维化的疗法,存在未满足的需求。
11.2.2癌症
12.tgf-β信号传导也与肿瘤进展有关,并且长期以来一直对抑制tgf-β途径作为癌症疗法感兴趣(syed,2016,j cell biochem.117(6):1279-87)。然而,由于对宿主毒性的担忧,因为tgf-β受体无处不在,并且担心会无意中促进肿瘤生长,因此大多数tgf-β抑制剂仍处于临床前发现阶段。
13.tgf-β由肿瘤细胞、癌症相关成纤维细胞(caf)和/或周围的肿瘤微环境(tme)细胞分泌。在tme的基质细胞中,caf是最丰富的,并且与癌症进展密切相关(pure and blomberg,2018,oncogene,37(32):4343-4357;calon et al.,2014,seminars in cancer bio,25:15-22;chen and song,2019,nat rev drug disc.18:90)。tgf-β是caf活化、募集和活力的关键驱动因子,tgf-β经由上皮间质转化(emt)驱动caf从组织驻留成纤维细胞和上皮细胞的分化并支持其生存。继而,caf对肿瘤生长、血管生成、癌症干性、ecm重塑、组织侵袭、转移甚至化学抗性都有影响(harryvan and van der burg,2019,j clin med,8:1989)。caf是复杂且通常是异质的细胞群,使用各种细胞内和细胞表面标志物的组合来识别caf,所述细胞内和细胞表面标志物包括细胞内α-平滑肌肌动蛋白(sma)和细胞表面成纤维细胞活化蛋白(fap)的升高表达(pure and blomberg,2018,oncogene,37(32):4343-4357)。在膀胱癌和结直肠癌患者中,tgf-β信号传导可促进免疫排除或“冷”肿瘤,其中caf将t细胞保持困在肿瘤外,从而在物理上阻止它们浸润肿瘤(hegde,2020,immunity,52:17-35;gajewski,2015,semin oncol,42:663-671;mariathasan and powles,2018,nature,554:544-48)。
14.尽管tgf-β疗法是治疗癌症感兴趣的,但由于tgf-β及其受体的广泛表达以及它们在组织(包括心脏和骨)的发育、维持和稳态中的作用,它们在历史上还没有达到其全部治疗潜力。此外,tgf-β是早期肿瘤抑制因子(suppressor),其负责控制早期肿瘤的生长,已经证明全身性tgf-β疗法会引起组织毒性并增加早期肿瘤生长(anderton and heier,2011,toxicologic path,39:916;stauber et al.,2014,clinical tox,4(3):1-10;lonning and mcpherson,2011,curr pharma biotech,12:2176-89)。
15.因此,需要将tgf-β抑制剂靶向至在tgf-β信号传导的抑制在治疗上有用的细胞类型,例如癌症相关成纤维细胞(“caf”),同时最小化宿主组织毒性。
16.3.发明概述
17.本公开涉及用于治疗纤维化和癌症的组合物和方法。通过将tgf-β抑制剂主要且优选仅靶向那些在其中将赋予tgf-β抑制剂治疗益处的那些细胞从而避免多效脱靶效应,组合物和方法有利地避免了与全身施用tgf-β抑制剂相关的中靶宿主毒性。
18.具体地,该组合物和方法经由与靶细胞细胞表面分子结合的靶向部分,例如抗体
或抗体片段将alk5抑制剂引导至肌成纤维细胞、活化的成纤维细胞(例如,癌症相关成纤维细胞(“caf”))和向肌成纤维细胞转变的成纤维细胞(每种细胞类型为“靶细胞”)。不受理论束缚,认为靶向部分的使用可导致alk5抑制剂定位于靶细胞并内化到靶细胞中,从而抑制靶细胞中的tgfβ途径,同时限制全身毒性。抑制例如肌成纤维细胞或向肌成纤维细胞转变的成纤维细胞中的tgfβ途径可导致对纤维发生的抑制(在患有纤维化或与纤维化相关的疾病的受试者的情况下)。caf中tgfβ途径的抑制可导致肿瘤进展的抑制(在受试者患有癌症的情况下)。不受理论束缚,认为选择性阻断caf中的tgf-β信号传导可以1)消除caf介导的对免疫细胞浸润的阻断,和/或2)驱动肿瘤清除,和/或3)降低caf活力和/或4)与全身性tgf-β抑制剂相关的旁路毒性问题。
19.因此,本公开提供靶向性药物缀合物(tdc),其中药物是alk5抑制剂。本公开的tdc包含靶向组分,例如结合靶细胞的细胞表面分子(例如,人肌成纤维细胞表面分子)的抗体或抗体片段。或者,靶向部分可包含基于非免疫球蛋白的肽或多肽,其与靶细胞表面分子的细胞表面结合。不受理论束缚,认为本公开的tdc可以通过促进靶细胞去分化为静止成纤维细胞和/或通过促进靶细胞凋亡来提供治疗效果。第5.2节描述了可用于本公开的tdc的示例性靶向部分。在一些实施方案中,alk5抑制剂是咪唑-苯并二氧杂环戊烯(imidazole-benzodioxol)化合物、咪唑-喹喔啉(imidazole-quinoxaline)化合物、吡唑-吡咯并(pyrazole-pyrrolo)化合物或噻唑(thiazole)类化合物。示例性alk5抑制剂在第5.3节和表1-3中进行了描述。在一些实施方案中,alk5抑制剂是n-甲基-2-(4-(4-(3-(6-甲基吡啶-2-基)-1h-吡唑-4-基)吡啶-2-基)苯氧基)乙烷-1-胺(本文称为“化合物c”)。
20.alk5抑制剂可以直接缀合至靶向部分或通过接头连接至靶向部分。接头可以是不可切割接头,或优选地,是可切割的接头。示例性的不可切割和可切割接头在第5.4节中描述。每个靶向部分连接的alk5抑制剂分子的平均数可以变化,并且通常每个靶向部分在2至8个alk5抑制剂分子的范围内。第5.5节详细描述了载药量。
21.本公开进一步提供了包含本公开的tdc的药物组合物。示例性药物赋形剂可用于配制包含本公开的tdc的药物组合物,该示例性药物赋形剂在第5.6节中描述。
22.本公开进一步提供通过向有需要的受试者施用本公开的tdc或本公开的药物组合物来治疗纤维化的方法和治疗癌症的方法。本公开的tdc和药物组合物可以作为单一疗法或作为组合疗法的一部分施用,例如与另一种治疗剂,如吡非尼酮或尼达尼布(当治疗患有纤维化或与纤维化相关的疾病的受试者时)或化疗剂(当治疗患有癌症的受试者时)组合施用。作为另一个实例,当治疗患有癌症的受试者时,tdc和药物组合物可以与检查点抑制剂组合施用。可以用本公开的tdc和药物组合物以及示例性组合疗法治疗的示例性病况类型在第5.7节中描述。
4.附图说明
23.图1a-1d显示了化合物a-d对hek293t细胞中tgf-β诱导的萤光素酶活性的抑制。图1a:化合物a;图1b:化合物b;图1c:化合物c;图1d:化合物d。
24.图2a-2c显示只有当转染人fap cdna并在细胞表面表达时,抗fap抗体才与hek细胞结合。图2a:未染色的hek细胞。图2b:用抗fap抗体染色的hek细胞。图2c:用fap cdna转染并用抗fap抗体染色的hek细胞。
25.图3a-3b显示了在靶向性药物缀合物syn-301(图3a)和syn-302(图3b)中使用的接头和有效负载。
26.图4a-4b显示了syn-301在表达人fap蛋白的hek细胞中抑制tgf-β信号传导。图4a:表达人fap蛋白的hek细胞中的相对萤光素酶报告基因表达。图4b:未转染的hek细胞中的相对萤光素酶报告基因表达。
27.图5a-5e显示了50-60%的wi-38细胞表达fap。图5a:未染色的wi-38细胞。图5b:用抗fap抗体染色的wi-38细胞。图5c:用syn-301染色的wi-38细胞。图5d:用syn-302染色的wi-38细胞。图5e:用同种型对照adc染色的wi-38细胞。
28.图6显示了由抗fap抗体(63%)、syn-301(63%)和syn-302(52%)诱导的fap内化百分比。
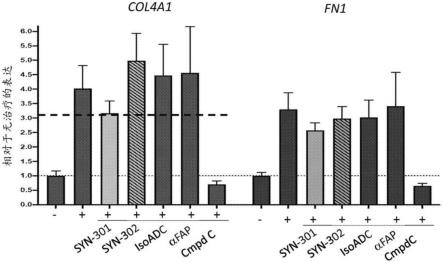
29.图7a-7b显示了syn-301和syn-302对wi-38细胞中胶原蛋白和纤连蛋白(图7a)和lrrc15(图7b)的rna表达的影响。
30.5.发明详述
31.本公开提供了可用于治疗纤维化和癌症的靶向性药物缀合物(tdc),其包含直接或通过接头与alk5抑制剂共价结合的靶向部分。第5.1节介绍了本公开的tdc的概述。tdc的靶向部分可以包括例如完整抗体或其片段。可以在本公开的tdc中使用的靶向部分在第5.2节中详细描述。可用于本公开的tdc的alk5抑制剂在第5.3节中进行了描述。本公开的tdc通常含有靶向部分和alk5抑制剂之间的接头。可用于本公开的tdc的示例性接头在第5.4节中进行了描述。本公开的tdc可以含有每个靶向部分不同数量的alk5抑制剂部分。第5.5节详细讨论了载药量。本公开进一步提供了包含本公开的tdc的药物制剂。包含tdc的药物制剂在第5.6节中进行了描述。本公开进一步提供了使用本公开的tdc治疗纤维化的方法和治疗癌症的方法。使用本公开的tdc作为单一疗法或作为组合疗法的一部分用于治疗纤维化或癌症的方法在第5.7节中描述。
32.5.1.药物偶联物
33.本公开的tdc通常由alk5抑制剂组成,所述alk5抑制剂通常经由接头共价连接至靶向部分(如抗体或抗体片段),使得共价连接不干扰与靶向部分的靶物的结合。
34.用于将药物缀合至靶向部分,如抗体和抗体片段的技术在本领域中是众所周知的(参见,例如,hellstrom et al.,controlled drug delivery,2nd ed.,at pp.623-53(robinson et al.,eds.,1987);thorpe et al.,1982,immunol.rev.62:119-58;dubowchik et al.,1999,pharmacology and therapeutics 83:67-123;和zhou,2017,biomedicines 5(4):e64))。alk5抑制剂优选经由位点特异性缀合与本公开的tdc中的靶向部分连接。例如,alk5抑制剂可以经由以下与靶向部分缀合:一种或多种天然或工程化的半胱氨酸、赖氨酸或谷氨酰胺残基、一种或多种非天然氨基酸(例如,对乙酰苯丙氨酸(pacf)、对叠氮基甲基-l-苯丙氨酸(pamf)或硒半胱氨酸(sec))、一种或多种聚糖(例如岩藻糖、6-硫代岩藻糖、半乳糖、n-乙酰半乳糖胺(galnac)、n-乙酰葡糖胺(glcnac)或唾液酸(sa))、或一个或多个四至六个氨基酸的短肽标签。参见,例如,zhou,2017,biomedicines 5(4):e64,其内容通过引用整体并入本文。
35.在一个实例中,靶向部分经由共价键(例如肽键)、通过靶向部分的n-端或c-端或在内部融合到另一种蛋白质(或其部分;例如蛋白质的至少10、20或50个氨基酸的部分)的
氨基酸序列。靶向部分可以在n端与其他蛋白质连接,例如抗体或抗体片段可以在抗体恒定域的n端连接。重组dna程序可用于产生此类融合,例如如wo 86/01533和ep0392745中所述。在另一个实例中,效应分子可以增加体内半衰期,和/或增强tdc向靶细胞的递送。这种类型的合适的效应分子的实例包括聚合物、白蛋白、白蛋白结合蛋白或白蛋白结合化合物,如在pct公开wo 2005/117984号中描述的那些。
36.代谢过程或反应可以是酶促过程,如tdc的肽接头的蛋白水解切割,或如腙、酯或酰胺的官能团的水解。细胞内代谢物包括但不限于肽和游离药物,它们在进入、扩散、摄取或转运进入细胞后经历了细胞内切割。
37.术语“细胞内切割的”和“细胞内切切割”是指药物缀合物上细胞内的代谢过程或反应,由此破坏了药物部分(d)和靶向部分之间的共价连接,例如接头,导致在细胞内从靶向部分解离的游离药物。因此,tdc的切割部分是细胞内代谢物。
38.5.2.靶向部分
39.本公开提供了药物缀合物,其中靶向部分与靶细胞细胞表面分子结合。靶向部分通常包含抗体或抗体片段(此类缀合物在本文中有时称为“抗体药物缀合物”或“adc”)。或者,靶向部分可以是基于非免疫球蛋白的,例如基于非免疫球蛋白的肽或多肽(例如,在靶细胞表面上表达的受体的配体)。因此,应当理解术语“靶向部分”涵盖肽(例如,长度为十至四十个氨基酸的肽)、单链多肽(例如,长度超过四十个氨基酸的多肽,例如单链可变区或scfv),以及包含多条多肽链的分子(例如,多聚体免疫球蛋白分子)。
40.除非另有说明,否则术语“抗体”是指特异性结合特定抗原或与特定抗原发生免疫反应的免疫球蛋白分子,其包括多克隆、单克隆、基因工程化的和其他修饰形式的抗体,包括但不限于嵌合抗体、人源化抗体、异源缀合抗体(例如双特异性抗体、双抗体、三抗体和四抗体)和抗体的抗体片段,包括例如fab'、f(ab')2、fab、fv、rigg和scfv片段。此外,除非另有说明,否则术语“单克隆抗体”(mab)既包括完整分子,也包括抗体片段(如,例如fab和f(ab')2片段),其可以与蛋白质特异性结合。fab和f(ab')2片段缺乏完整抗体的fc片段,从动物或植物的循环中清除得更快,并且可能比完整抗体具有更少的非特异性组织结合(wahl et al.,1983,j.nucl.med.24:316)。
41.提及“vh”是指抗体的免疫球蛋白重链的可变区,其包括fv、scfv或fab的重链。提及“vl”是指免疫球蛋白轻链的可变区,其包括fv、scfv、dsfv或fab的轻链。抗体和免疫球蛋白(ig)是具有相同结构特征的糖蛋白。虽然抗体表现出对特定靶的结合特异性,但免疫球蛋白包括抗体和其他缺乏靶特异性的抗体样分子。天然抗体和免疫球蛋白通常是约150,000道尔顿的异四聚体糖蛋白,由两条相同的轻(l)链和两条相同的重(h)链组成。每条重链在氨基端都有可变域(vh),然后是多个恒定域。每条轻链在氨基端(vl)具有可变域,在羧基端具有恒定域。
42.对于细胞内alk5抑制剂的最佳递送,靶向部分优选内化性的,例如内化性抗体。在内化性靶向部分与细胞表面上的靶分子结合后,细胞由于结合而内化内化性靶向部分。这样做的效果是细胞摄入tdc。允许确定内化,例如在与其抗原结合后的抗体的内化的方法是本领域技术人员已知的并且描述于例如pct公开wo 2007/070538号的第80页中。一旦内化,如果使用可切割的接头将alk5抑制剂连接到靶向部分,例如第5.4节中所述,则alk5抑制剂可以通过溶酶体中的切割或通过其他细胞机制从靶向部分释放。
43.术语“抗体片段”是指全长抗体的一部分,通常是靶结合区或可变区。抗体片段的实例包括fab片段、fab'片段、f(ab')2片段、fv片段、scfv片段、dsfv片段或单域抗体。
[0044]“fv”片段是含有完整靶识别和结合位点的最小抗体片段。该区域由一个重链和一个轻链可变域的二聚体组成,形成紧密的非共价结合(vh-vl二聚体)。正是在这种配置中,每个可变域的三个cdr相互作用以在vh-vl二聚体表面上定义靶结合位点。通常,六个cdr赋予抗体靶结合特异性。然而,在某些情况下,甚至单个可变域(或仅包含三个对靶特异的cdr的fv的一半)也可以具有识别和结合靶的能力。
[0045]“单链fv”或“scfv”抗体片段包含单个多肽链中抗体的vh和vl域。通常,scfv多肽进一步包含位于vh和vl域之间的多肽接头,其使scfv能够形成靶结合所需的结构。本领域已经描述了各种scfv接头。参见,例如,shen et al.,2008,anal chem.80(6):1910
–
1917;yusakul,et al.,2016,biosci biotechnol biochem.80(7):1306-12。示例性scfv接头包含序列(ggggs)n,其中n在1和10之间。
[0046]“二硫化物稳定的fv”或“dsfv”抗体片段包含通过域间二硫化物稳定的抗体的vh和vl域。参见brinkmann u.,2010,disulfide-stabilized fv fragments.in:kontermann r.,d
ü
bel s.(eds)antibody engineering.springer,berlin,heidelberg。
[0047]“单域抗体”由对靶(例如,fap)表现出足够亲和力的单个vh或vl域(例如,人或鼠抗体的)组成。在具体实施方案中,单域抗体是骆驼vhh抗体片段(参见,例如riechmann,1999,journal of immunological methods 231:25-38)。在本公开的tdc中使用单域抗体可能是有利的,因为它们与全长抗体相比尺寸小、溶解度高、稳定性高和体内组织穿透性优异。已经描述了制备单域抗体的各种方法。参见例如,美国专利10,030,068号、us 2006/0246058、美国专利7,371,849号、vincke et al,2008,jbc,284(5):3273-3284。
[0048]
fab片段含有轻链的恒定域和重链的第一恒定域(ch1)。fab'片段与fab片段的不同之处在于在重链ch1域的羧基端添加了几个残基,包括来自抗体铰链区的一个或多个半胱氨酸。f(ab')片段是通过切割f(ab')2胃蛋白酶消化产物的铰链半胱氨酸处的二硫键而产生的。本领域普通技术人员已知抗体片段的额外化学偶联。
[0049]
在某些实施方案中,本公开的抗体是单克隆抗体。如本文所用,术语“单克隆抗体”不限于通过杂交瘤技术产生的抗体。术语“单克隆抗体”是指衍生自单个克隆的抗体,单个克隆包括任何真核、原核或噬菌体克隆,而不是其生产方法。可使用本领域已知的多种技术制备与本公开有关的单克隆抗体,所述多种技术包括使用杂交瘤、重组和噬菌体展示技术或其组合。本公开的抗体包括嵌合的、灵长类化的、人源化的或人抗体。
[0050]
本发明的抗体可以是嵌合抗体。如本文所用,术语“嵌合”抗体是指具具有衍生自非人免疫球蛋白(如大鼠或小鼠抗体)的可变序列和通常选自人免疫球蛋白模板的人免疫球蛋白恒定区的抗体。产生嵌合抗体的方法是本领域已知的。参见,例如,morrison,1985,science 229(4719):1202-7;oi et al.,1986,biotechniques 4:214-221;gillies et al.,1985,j.immunol.methods 125:191-202;美国专利5,807,715号;4,816,567号和4,816,397号,其通过引用整体并入本文。
[0051]
本公开的抗体可以是人源化的。非人(例如鼠)抗体的“人源化”形式是嵌合免疫球蛋白、免疫球蛋白链或其片段(如fv、fab、fab'、f(ab')2或抗体的其他靶结合亚域),它们含有衍生自非人免疫球蛋白的最小序列。一般而言,人源化抗体将含有基本上所有、至少一
个、通常是两个可变域,其中所有或基本上所有的cdr区对应于非人免疫球蛋白的cdr区和所有或基本上所有的fr区是人免疫球蛋白序列的那些。人源化抗体还可以包含免疫球蛋白恒定区(fc)的至少一部分,通常是人免疫球蛋白共有序列的一部分。抗体人源化的方法是本领域已知的。参见,例如,riechmann et al.,1988,nature 332:323-7;美国专利5,530,101号;5,585,089号;5,693,761号;5,693,762号;和6,180,370号;queen et al.;欧洲专利公开ep239400号;pct公开wo 91/09967号;美国专利5,225,539号;欧洲专利公开ep592106号;欧洲专利公开ep519596号;padlan,1991,mol.immunol.,28:489-498;studnicka et al.,1994,prot.eng.7:805-814;roguska et al.,1994,proc.natl.acad.sci.91:969-973;和美国专利5,565,332号,所有这些都通过引用整体并入本文。
[0052]
本发明的抗体可以是人抗体。对于人患者的治疗性治疗,可能需要完全“人”抗体。如本文所用,“人抗体”包括具有人免疫球蛋白的氨基酸序列的抗体,并且包括从人免疫球蛋白文库或从一种或多种人免疫球蛋白转基因且不表达内源性免疫球蛋白的动物分离的抗体。人抗体可以通过本领域已知的多种方法制成,该方法包括使用衍生自人免疫球蛋白序列的抗体文库的噬菌体展示方法。参见美国专利4,444,887号和4,716,111号;和pct公开wo 98/46645号;wo 98/50433号;wo 98/24893号;wo 98/16654号;wo 96/34096号;wo 96/33735号;和wo 91/10741号,每一个都通过引用整体并入本文。也可以使用不能表达功能性内源性免疫球蛋白但可以表达人免疫球蛋白基因的转基因小鼠来产生人抗体。参见例如pct公开wo 98/24893号;wo 92/01047号;wo 96/34096号;wo 96/33735号;美国专利5,413,923号;5,625,126号;5,633,425号;5,569,825号;5,661,016号;5,545,806号;5,814,318号;5,885,793号;5,916,771号;5,939,598号和5,939,598号,其通过引用整体并入本文。此外,公司如medarex(princeton,n.j.)、astellas pharma(deerfield,ill.)、amgen(thousand oaks,calif.)和regeneron(tarrytown,n.y.)可使用与上述类似的技术参与以提供针对选定抗原的人抗体。可以使用称为“引导选择”的技术生成识别选定表位的完全人抗体。在该方法中,使用选择的非人单克隆抗体,例如小鼠抗体来引导选择识别相同表位的完全人抗体(jespers et al.,1988,biotechnology 12:899-903)。
[0053]
本公开的抗体可以是灵长类化的。术语“灵长类化抗体”是指包含猴可变区和人恒定区的抗体。产生灵长类化抗体的方法是本领域已知的。参见,例如,美国专利5,658,570号;5,681,722号;5,693,780号和5,693,780号,其通过引用整体并入本文。
[0054]
本公开的抗体包括衍生化抗体。例如,但不限于,衍生化抗体通常通过糖基化、乙酰化、聚乙二醇化、磷酸化、酰胺化、通过已知保护/阻断基团衍生、蛋白水解切割、与细胞配体或其他蛋白质的连接等进行修饰(参见第5.1节抗体缀合物的讨论)。许多化学修饰中的任何一种都可以通过已知技术进行,包括但不限于特异性化学切割、乙酰化、甲酰化、衣霉素的代谢合成等。另外,衍生物可以含有一种或多种非天然氨基酸,例如,使用ambrx技术(参见,例如,wolfson,2006,chem.biol.13(10):1011-2)。
[0055]
在本公开的另一个实施方案中,抗体或其片段可以是如下的抗体或抗体片段,其序列已经经过修饰以相对于相应的野生型序列改变至少一个恒定区介导的生物效应子功能。例如,在一些实施方案中,可以修饰本公开的抗体以相对于未修饰的抗体减少至少一种恒定区介导的生物效应子功能,例如减少与fc受体(fcγr)或clq的结合。可以通过在fcγr或c1q相互作用所必需的特定区域突变抗体的免疫球蛋白恒定区片段来减少fcγr和c1q结
合(参见,例如,canfield and morrison,1991,j.exp.med.173:1483-1491;lund et al.,1991,j.immunol.147:2657-2662;lo.et al.,2017,j biol chem 292:3900-08;wang et al.,2018,protein cell 9:63-73)。
[0056]
抗体与fcγr结合能力的降低还可以降低依赖fcγr相互作用的其他效应子功能,如调理作用、吞噬作用和抗体依赖性细胞毒性(“adcc”),而c1q结合能力的减少可以减少补体依赖性细胞毒性(“cdcc”)。因此,效应子功能的减少或消除可以防止经由adcc或cdcc对由本公开的药物缀合物靶向的靶细胞的破坏。因此,在一些实施方案中,通过抗体的fc部分的选择性突变修饰抗体的效应子功能,以使其保持抗原特异性和内化能力但消除adcc/cdcc功能。在其他实施方案中,未修饰抗体的效应子功能以减少或消除adcc/cdcc功能。不受理论束缚,认为本发明包含有adcc/cdcc功能的抗体或抗体片段的tdc可以通过促进靶细胞凋亡来增强抑制tgfβ信号传导的治疗效果,从而进一步抑制纤维发生。
[0057]
本领域已经描述了许多突变以减少fcγr和clq结合,并且此类突变可以包括在本公开的药物缀合物中。例如,美国专利6,737,056号公开了在位置238、265、269、270、292、294、295、298、303、324、327、329、333、335、338、373、376、414、416、419、435、438或439处的单位置fc区氨基酸修饰,导致与fcγrii和fcγrii的结合减少。美国专利9,790,268号公开了氨基酸位置298处的天冬酰胺残基和氨基酸位置300处的丝氨酸或苏氨酸残基减少了fcγr结合。pct公开wo 2014/190441号描述了有降低的fcγr结合的修饰的fc域具有l234d/l235e:l234r/l235r/e233k、l234d/l235e/d265s:e233k/l234r/l235r/d265s、l234d/l235e/e269k:e233k/l234r/l235r/e269k、l234d/l235e/k322a:e233k/l234r/l235r/k322a、l234d/l235e/p329w:e233k/l234r/l235r/p329w、l234d/l235e/e269k/d265s/k322a:e233k/l234r/l235r/e269k/d265s/k322a、
[0058]
l234d/l235e/e269k/d265s/k322e/e333k:e233k/l234r/l235r/e269k/d265s/k322e/e333k突变,其中分号之前的一组突变在第一fc多肽中,分号之后的突变在fc二聚体的第二个fc多肽中。
[0059]
可以减少fcγr受体结合以及c1q结合的突变包括n297a、n297q、n297g、d265a/n297a、d265a/n297g、l235e、l234a/l235a和l234a/l235a/p329a(lo.et al.,2017,j biol chem 292:3900-08;wang et al.,2018,protein cell 9:63-73)。
[0060]
作为突变恒定区以减少效应子功能,例如,如上所述突变fc域的替代,可以通过利用抗体片段(例如,fab、fab'或f(ab')2)来消除效应子功能。
[0061]
在本公开的其他实施方案中,可以修饰抗体或其片段以相对于未修饰的抗体获得或改善至少一种恒定区介导的生物效应子功能,例如,以增强fcγr相互作用(参见,例如,us 2006/0134709)。例如,本公开的抗体可以具有以比对应的野生型恒定区更高的亲和力,结合fcγriia、fcγriib和/或fcγriiia的恒定区。
[0062]
因此,本公开的抗体可以具有导致调理作用、吞噬作用或adcc减少的生物活性的改变。此类改变在本领域中是已知的。例如,减少adcc活性的抗体修饰描述于美国专利5,834,597号。
[0063]
在又一方面,抗体或其片段可以是已修饰以增加或减少它们对胎儿fc受体fcrn的结合亲和力的抗体或抗体片段,例如,通过突变涉及fcrn相互作用的特定区域的免疫球蛋白恒定区片段(参见例如wo 2005/123780)。此类突变可以增加抗体与fcrn的结合,从而保
护抗体免于降解并增加其半衰期。
[0064]
在其他方面,抗体具有插入其一个或多个高变区的一个或多个氨基酸,例如,如jung and pl
ü
ckthun,1997,protein engineering 10(9):959-966;yazaki et al.,2004,protein eng.des sel.17(5):481-9;和美国专利公开2007/0280931号中描述的。
[0065]
靶向部分的靶物将取决于tdc的所需治疗应用。通常,靶物是存在于希望接受alk5抑制剂递送的细胞的表面上的分子,如肌成纤维细胞或癌症相关的成纤维细胞,并且靶向部分优选在与靶物结合后内化。内化性靶向部分,例如抗体,描述于例如franke et al.,2000,cancer biother.radiopharm.15:459 76;murray,2000,semin.oncol.27:64 70;breitling et al.,recombinant antibodies,john wiley,and sons,new york,1998)。在某些实施方案中,靶向部分不显著阻断靶细胞表面分子的活性。例如,激动性抗体或其片段或非拮抗性抗体或其片段可以用作靶向部分,例如当靶分子是fap或αvβ6时。
[0066]
优选地,靶向部分选择性地结合肌成纤维细胞、活化的成纤维细胞、向肌成纤维细胞转变的成纤维细胞或其组合,而不是结合其他细胞类型,例如静止成纤维细胞、肺上皮细胞、肝细胞、t细胞、不表达胶原蛋白的细胞和/或不表达α-平滑肌肌动蛋白(αsma)的细胞。静止或沉寂的(quiescent)成纤维细胞可鉴定为梭形单细胞,而活化的成纤维细胞获得αsma和波形蛋白(vimentin)的表达并变成星状。选择性结合可以通过靶向在一个或多个靶细胞表面上表达但在其他细胞类型上表达低或不表达的细胞表面分子来实现。选择性可以通过本领域已知的各种测定来测量,例如通过流式细胞术。在一些实施方案中,相对于静止成纤维细胞,本公开的tdc的靶向部分对肌成纤维细胞具有至少2倍或至少3倍的选择性,例如通过facs测量的(例如,2至1000倍、2至100倍、2至50倍、2至10倍、3至1000倍、3至100倍、3至50倍、3至10倍、5至1000倍、5至100倍、5至50倍、5至10倍、20至1000倍、20至100倍、20至50倍、50至1000倍、50至100倍、100至1000倍或超过1000倍)。在一些实施方案中,相对于静止的成纤维细胞,本公开的tdc的靶向部分对活化的成纤维细胞(例如,caf)具有至少2倍或至少3倍的选择性,例如通过facs测量的(例如,2至1000倍、2至100倍、2至50倍、2至10倍、3至1000倍、3至100倍、3至50倍、3至10倍、5至1000倍、5至100倍、5至50倍、5至10倍、20至1000倍、20至100倍、20至50倍、50至1000倍、50至100倍、100至1000倍或超过1000倍)。在一些实施方案中,相对于静止的成纤维细胞,本公开的tdc的靶向部分对于向肌成纤维细胞转变的成纤维细胞具有至少2倍或至少3倍的选择性,例如通过facs测量的(例如,2至1000倍、2至100倍、2至50倍、2至10倍、3至1000倍、3至100倍、3至50倍、3至10倍、5至1000倍、5至100倍、5至50倍、5至10倍、20至1000倍、20至100倍、20至50倍、50至1000倍、50至100倍、100至1000倍或超过1000倍)。适合通过靶向部分靶向的细胞表面分子的实例包括但不限于成纤维细胞活化蛋白(fap)、血小板衍生的生长因子受体β(pdgfr-β)、成纤维细胞生长因子受体1(fgfr1)、过氧化物酶体增殖物激活受体γ(ppar-γ)、成纤维细胞特异性蛋白1(fsp1)、胶质纤维酸性蛋白(gfap)、肌成束蛋白(fascin)、cd147、c-x-c趋化因子受体4型(cxcr4)、αvβ6、axl、和mertk。axl和mertk是tam受体激酶家族的成员。适合通过靶向部分靶向的细胞表面分子的进一步实例是富含亮氨酸重复15(lrrc15)。
[0067]
在一些实施方案中,本公开的tdc的靶向部分结合fap。在其他实施方案中,本公开的tdc的靶向部分结合pdgfr-β。在其他实施方案中,本公开的tdc的靶向部分结合fgfr1。在其他实施方案中,本公开的tdc的靶向部分结合ppar-γ。在其他实施方案中,本公开的tdc
的靶向部分结合fsp1。在其他实施方案中,本公开的tdc的靶向部分结合gfap。在其他实施方案中,本公开的tdc的靶向部分结合肌成束蛋白。在其他实施方案中,本公开的tdc的靶向部分结合cd147。在其他实施方案中,本公开的tdc的靶向部分结合cxcr4。在其他实施方式中,本公开的tdc的靶向部分结合αvβ6。在其他实施方式中,本公开的tdc的靶向部分结合axl。在其他实施方案中,本公开的tdc的靶向部分结合mertk。在其他实施方案中,本公开的tdc的靶向部分结合lrrc15。
[0068]
成纤维细胞活化蛋白(fap)是二肽基肽酶(dpp)家族的成员,以ii型整合膜蛋白的表达。膜结合fap含有短的细胞质尾(残基1-4)、跨膜区(残基5-25)和细胞外域(残基26-760)(www.uniprot.org/uniprot/q12884)。fap在细胞表面以170kd二聚体的形式具有活性,但其胞外域在从膜上切割后也具有活性。fap同时具有二肽酶和内肽酶活性。与dpp酶家族的其他成员一样,fap是脯氨酰特异性丝氨酸蛋白酶,但fap也具有明胶酶活性,这允许其降解变性胶原蛋白i和iii、人成纤维细胞生长因子21(fgf-21)和人α2抗纤溶酶。fap在发育过程中表达,但很少在健康成人组织中表达。然而,活化成纤维细胞中升高的fap表达在炎症和活化组织重塑部位,包括伤口愈合、纤维化和癌症中表达。由于fap是多种疾病环境中活化成纤维细胞和肌成纤维细胞的标志物,因此fap靶向疗法不仅限于癌症患者,而且广泛适用于治疗ipf和其他纤维化疾病,例如nash(肝)、心脏和/或肾纤维化。此外,tgf-β可以增加fap的表达,其中fap的启动子具有smad3结合元件。由于这种有限的组织表达和在纤维化组织中的局部表达,已将fap用于成像以及针对疾病组织的治疗靶向。在癌症中,fap在支持癌症生长和转移的癌症相关成纤维细胞(caf)上高度且选择性地表达。fap很容易内化到细胞中,使其成为出色的细胞靶向和递送媒介物。与细胞毒性有效载荷偶联的抗fap抗体与化疗组合表明了肿瘤清除,而与放射性核苷酸偶联的抗fap可提高体内鼠肿瘤模型的存活率(ostermann et al.,2008,clin cancer res,2008.14(14):4584-92;fang et al.,2016,int j cancer,138(4):1013-23;fischer et al.,clin cancer res 18(22):6208-18)。虽然sibroluzumab是非偶联人源化抗fap抗体,但在转移性fap 癌症患者中并未显示单药疗效,但它确实在肿瘤而非正常组织中特异性积聚,并且在有限不良事件的患者中具有良好的耐受性。fap的可溶形式,也称为抗纤溶酶切割酶(apce),其缺乏膜结合fap的细胞质尾和跨膜区。已显示可溶性fap在肝硬化患者中升高,其水平随着疾病的严重程度而增加(e willige et al.,2013,j thromb haemost.11(11):2029-36)。优选地,靶向fap的靶向部分优先结合fap的膜结合形式而不是可溶形式。不受理论的束缚,认为可溶性fap可以充当沉淀物,与优先结合膜结合形式fap的tdc比与可溶形式fap结合的tdc的体内活性减少。
[0069]
结合fap的抗体的实例描述于wo 2012/020006、wo 2016/116399(例如抗体f5)和wo 2016/110598,其内容通过引用整体并入本文。在一些实施方案中,本公开的tdc的靶向部分包含在wo 2012/020006、wo 2016/116399或wo 2016/110598中描述的抗fap抗体或其片段(例如,fab片段、fab'片段、f(ab')2片段、fv片段、scfv片段、dsfv片段或单域抗体)。在一些实施方案中,本公开的tdc的靶向部分包含sibroluzumab(boehringer ingelheim)或其片段(例如,fab片段、fab'片段、f(ab')2片段、fv片段、scfv片段、dsfv片段或单域抗体)。
[0070]
结合pdgfr-β的抗体的实例描述于wo 2017/106609和wo 2014/109999,其内容通过引用整体并入本文。在一些实施方案中,本公开的tdc的靶向部分包含wo 2017/106609或wo 2014/109999中描述的抗pdgfr-β抗体或其片段(例如,fab片段、fab'片段、f(ab')2片
段、fv片段、scfv片段、dsfv片段或单域抗体)。在一些实施方案中,本公开的tdc的靶向部分包含imc-2c5(imclone)或其片段(例如,例如,fab片段、fab'片段、f(ab')2片段、fv片段、scfv片段、dsfv片段或单域抗体)。
[0071]
结合fgfr1的抗体的实例描述于wo 2018/095932和wo 2012/125124,其内容通过引用整体并入本文。在一些实施方案中,本公开的tdc的靶向部分包含在wo 2018/095932或wo 2012/125124中描述的抗fgfr1抗体或其片段(例如,fab片段、fab'片段、f(ab')2片段、fv片段、scfv片段、dsfv片段或单域抗体)。在一些实施方案中,本公开的tdc的靶向部分包含抗体,该抗体包含具有wo 2012/125124的seq id no:45的氨基酸序列的重链和具有wo 2012/125124s的eq id no:50的氨基酸序列的轻链或其片段(例如,fab片段、fab'片段、f(ab')2片段、fv片段、scfv片段、dsfv片段或单域抗体)的描述。
[0072]
结合ppar-γ的抗体的实例描述于wo 2005/026336中,其内容通过引用整体并入本文。在一些实施方案中,本公开的tdc的靶向部分包含在wo 2005/026336中描述的抗ppar-γ抗体或其片段(例如,fab片段、fab'片段、f(ab')2片段、fv片段、scfv片段、dsfv片段或单域抗体)。在一些实施方案中,本公开的tdc的靶向部分包含在wo 2005/026336中指定为pγ48.34a的抗体或其片段(例如,fab片段、fab'片段、f(ab')2片段、fv片段、scfv片段、dsfv片段或单域抗体)。
[0073]
结合fsp1的抗体的实例描述于wo 2011/157724中,其内容通过引用以其整体并入本文。在一些实施方案中,本公开的tdc的靶向部分包含wo 2011/157724中描述的抗fsp1抗体或其片段(例如,fab片段、fab'片段、f(ab')2片段、fv片段、scfv片段、dsfv片段或单域抗体)。在一些实施方案中,本公开的tdc的靶向部分包含抗体mab4137(r&d systems)或其片段(例如,fab片段、fab'片段、f(ab')2片段、fv片段、scfv片段、dsfv片段或单域抗体)。
[0074]
结合gfap的抗体的实例描述于wo 2018/081649中,其内容通过引用以其整体并入本文。在一些实施方案中,本公开的tdc的靶向部分包含wo 2018/081649中描述的抗gfap抗体或其片段(例如,fab片段、fab'片段、f(ab')2片段、fv片段、scfv片段、dsfv片段或单域抗体)。在一些实施方案中,本公开的tdc的靶向部分包含在wo 2018/081649中指定为gfap-1至gfap-19的抗体之一或其片段(例如,fab片段、fab'片段、f(ab')2片段、fv片段、scfv片段、dsfv片段或单域抗体)。
[0075]
结合肌成束蛋白的抗体的实例包括fcn01(thermofisher)、ab126772(abcam)和ab183891(abcam)。在一些实施方案中,本公开的tdc的靶向部分包含fcn01、ab126772或ab183891之一或其片段(例如,fab片段、fab'片段、f(ab')2片段、fv片段、scfv片段、dsfv片段或单域抗体)。
[0076]
结合cd147的抗体的实例描述于wo 2015/160853、wo 2018/121578和wo 2018/165619,其内容通过引用整体并入本文。在一些实施方案中,本公开的tdc的靶向部分包含在wo 2015/160853、wo 2018/121578或wo 2018/165619中描述的抗cd147抗体或其片段(例如,fab片段、fab'片段、f(ab')2片段、fv片段、scfv片段、dsfv片段或单域抗体)。在一些实施方案中,本公开的tdc的靶向部分包含在wo 2018/165619中指定为3a11的抗体或wo 2018/165619中描述的人源化变体或此类抗体的片段(例如,fab片段,fab'片段、f(ab')2片段、fv片段、scfv片段、dsfv片段或单域抗体)。
[0077]
结合cxcr4的抗体的实例描述于wo 2011/098762、wo 2008/060367和wo 2006/
089141,其内容通过引用整体并入本文。在一些实施方案中,本公开的tdc的靶向部分包含在wo 2011/098762、wo 2008/060367或wo 2006/089141中描述的抗cxcr4抗体或其片段(例如,fab片段、fab'片段、f(ab')2片段、fv片段、scfv片段、dsfv片段或单域抗体)。在一些实施方案中,本公开的tdc的靶向部分包含wo 2011/098762中描述的抗体c-9p21、b-1m22、c-1i24、d-1k21或9n10或其片段(例如,fab片段,fab'片段、f(ab')2片段、fv片段、scfv片段、dsfv片段或单域抗体)。
[0078]
结合αvβ6的抗体的实例描述于wo 2008/112004和wo 2013/123152中,其内容通过引用整体并入本文。在一些实施方案中,本公开的tdc的靶向部分包含在wo 2008/112004或wo 2013/123152中描述的抗αvβ6抗体或其片段(例如,fab片段、fab'片段、f(ab')2片段、fv片段、scfv片段、dsfv片段或单域抗体)。在一些实施方案中,本公开的tdc的靶向部分包含抗体stx-100(biogen)或其片段(例如,fab片段、fab'片段、f(ab')2片段、fv片段、scfv片段、dsfv片段或单域抗体)。
[0079]
结合axl的抗体的实例描述于wo 2009/062690、wo 2010/130751、wo 2015/193430和wo 2016/005593,其内容通过引用整体并入本文。在一些实施方案中,本公开的tdc的靶向部分包含在wo 2009/062690、wo 2010/130751、wo 2015/193430或wo 2016/005593中描述的抗axl抗体,或其片段(例如,fab片段、fab'片段、f(ab')2片段、fv片段、scfv片段、dsfv片段或单域抗体)。在一些实施方案中,本公开的tdc的靶向部分包含adct-601(adc therapeutics)的抗体或其片段(例如,fab片段、fab'片段、f(ab')2片段、fv片段、scfv片段、dsfv片段或单域抗体)。
[0080]
结合mertk的抗体的实例描述于wo 2016/106221、wo 2019/005756和wo 2019/084307,其内容通过引用整体并入本文。在一些实施方案中,本公开的tdc的靶向部分包含在wo 2016/106221、wo 2019/005756或wo 2019/084307中描述的抗mertk抗体或其片段(例如,fab片段、fab'片段、f(ab')2片段、fv片段、scfv片段、dsfv片段或单域抗体)。在一些实施方案中,本公开的tdc的靶向部分包含抗体rgx-019(rgenix)或其片段(例如,fab片段、fab'片段、f(ab')2片段、fv片段、scfv片段、dsfv片段或单域抗体)。
[0081]
lrrc15在许多癌症(例如,乳腺癌、头颈癌、肺癌、胰腺癌、卵巢癌、结肠癌、肾脏癌、食道癌、胃腺癌和膀胱癌)中的癌症相关成纤维细胞上表达(purcell et al.,2018,cancer res.78(14):4059-4072;dominguez et al.,2019,cancer discovery 10(2):232-253)。因此,在一些实施方案中,本公开的tdc靶向lrrc15。结合lrrc15的抗体的实例描述于wo 2017/095805中,其内容通过引用整体并入本文。与lrrc15结合的抗体也是可商业获得的,例如abcam目录#ab150376和creative biolabs目录#tab-0709cl。abbv-085(abbvie)是含有mmae的针对lrrc15的adc(purcell et al.,2018,cancer res.78(14):4059-4072)。在一些实施方案中,本公开的tdc的靶向部分包含wo 2017/095805中描述的抗体、abbv-085的抗体、本段中描述的可商业获得的抗体之一、或其片段(例如,fab片段、fab'片段、f(ab')2片段、fv片段、scfv片段、dsfv片段或单域抗体)。
[0082]
5.3 alk5抑制剂
[0083]
本公开的alk5抑制剂优选地是小分子,该小分子竞争性地和可逆地结合到alk5受体的细胞质激酶域中的atp结合位点。
[0084]
alk5对alk5相对于其他tgf-β家族受体,如alk4和/或alk7和/或tgf-β受体ii,可
以但不需要是特异性和选择性的。在一些实施方案中,alk5抑制剂对alk5和tgf-β受体ii均具有活性。在一些实施方案中,alk5抑制剂对bmp ii受体具有有限的抑制活性。
[0085]
当在使用hek293t细胞的体外细胞测定中测量时,本公开的alk5抑制剂优选具有100nm或更小、更优选50nm或更小、并且最优选20nm或更小的ic
50
。下文第6.6节中阐述了示例性细胞测定。
[0086]
适用于本公开的tdc中的alk5抑制剂的阐释性实例包括咪唑-苯并二氧杂环戊烯化合物、咪唑-喹喔啉化合物、吡唑-吡咯并化合物和噻唑类化合物。
[0087]
根据本公开的一个方面,咪唑-苯并二氧杂环戊烯型alk5抑制剂具有下式
[0088][0089]
其中r1是氢或具有1至约5个碳原子的低级烷基,r2是氢或具有1至约5个碳原子的低级烷基并且r3是酰胺、腈、具有1至约3个碳原子的炔基、羧基或具有1至约5个碳原子的烷醇基团,a为直接键或具有1至约5个碳原子的烷基,b为直接键或具有1至约5个碳原子的烷基。在本公开的单独优选实施方案中,r2是氢或甲基,a具有1个碳原子并且b是与苄基基团的直接键并且r3是酰胺。在本公开的组合优选实施方案中,r2是氢或甲基,a具有1个碳原子且b是与苄基的直接键。
[0090]
根据本公开的另一方面,咪唑-喹喔啉型alk5抑制剂具有下式
[0091]
[0092]
其中r1是氢或具有1至约5个碳原子的低级烷基,r2是氢、卤素或具有1至约5个碳原子的低级烷基,r3是酰胺、腈、具有1至约3个碳原子的炔基,羧基或具有1至约5个碳原子的烷醇基团,a为直接键或具有1至约5个碳原子的烷基,b为直接键或具有1至约5个碳原子的烷基。在本公开的单独优选实施方案中,r2是氢或甲基,卤素包括氟或氯,a具有1个碳原子并且b是与苄基的直接键并且r3是酰胺。在本公开的组合优选实施方案中,r2是氢或甲基,a具有1个碳原子且b是与苄基基团的直接键。
[0093]
根据本公开的另一方面,吡唑型alk5抑制剂具有下式
[0094][0095]
其中r2是氢、卤素或具有1至约5个碳原子的低级烷基,r4是氢、卤素、具有1至约5个碳原子的低级烷基、具有1至约5个碳原子的烷氧基、卤代烷基、羧基、羧基烷基酯、腈、烷基胺或具有下式的基团
[0096][0097]
其中r5是具有1至约5个碳原子的低级烷基、卤素或吗啉代,并且r6是吡咯、环己基、吗啉代、吡唑、吡喃、咪唑、恶烷、吡咯烷基或烷基胺,并且a是直接键或具有1至约5个碳原子的烷基。
[0098]
根据本公开的另一个方面,吡唑-吡咯并型alk5抑制剂具有下式
[0099][0100]
其中r7是氢、卤素、具有1至约5个碳原子的低级烷基、烷醇、吗啉代或烷基胺,r2是氢、卤素或具有1至约5个碳原子的低级烷基并且r8是氢、羟基,氨基、卤素或具有下式的基团
[0101][0102]
其中r5是哌嗪基,r6是吗啉代、哌啶基、哌嗪基、烷氧基、羟基、恶烷、卤素、硫代烷基或烷基胺,并且a是具有1至约5个碳原子的低级烷基。
[0103]
根据本公开的另一方面,噻唑型alk5抑制剂具有下式
[0104][0105]
其中r9是氢、卤素或具有1至约5个碳原子的低级烷基,并且r
10
是氢或具有1至约5个碳原子的低级烷基。
[0106]
在某些实施方案中,alk5抑制剂选自下表1中指定为a至n的任何化合物:
[0107]
[0108]
[0109]
[0110][0111]
在进一步的具体实施方案中,alk5抑制剂选自下表2中指定为1至283的任何化合物:
[0112]
[0113]
[0114]
[0115]
[0116]
[0117]
[0118]
[0119]
[0120]
[0121]
[0122]
[0123]
[0124]
[0125]
[0126][0127]
alk5抑制剂的制备和使用是众所周知的,并且在科学和专利文献中有充分的记载。pct公开wo 2000/61576号和美国专利公开us 2003/0149277号公开了三芳基咪唑衍生物及其作为alk5抑制剂的用途。pct公开wo 2001/62756号公开了吡啶基咪唑衍生物及其作为alk5抑制剂的用途。pct公开wo 2002/055077号公开了咪唑基环状缩醛衍生物作为alk5抑制剂的用途。pct公开wo 2003/087304号公开了三取代杂芳基及其作为alk5和/或alk4抑制剂的用途。wo 2005/103028,美国专利公开us 2008/0319012号和美国专利7,407,958号公开了2-吡啶基取代的咪唑作为alk5和/或alk4抑制剂。代表性化合物之一,in-1130,在几种动物模型中显示出alk5和/或alk4抑制剂活性。以下专利和专利出版物提供了alk5抑制剂的额外实例,并提供了阐释性的合成方案和使用alk5抑制剂的方法:美国专利6,465,493;6,906,089;7,365,066;7,087,626;7,368,445;7,265,225;7,405,299;7,407,958;7,511,056;7,612,094;7,691,865;7,863,288;8,410,146;8,410,146;8,420,685;8,513,2228,614,226;8,791,113;8,815,893;8,846,931;8,912,216;8,987,301;9,051,307;9,051,318;9,073,918号和pct公开wo 2004/065392;wo 2009/050183;wo 2009/133070;wo 2011/146287;和wo 2013/009140号。前述专利和专利出版物通过引用整体并入。
[0128]
几种alk5抑制剂是可商业获得的,其包括sb-525334(cas 356559-20-1)、sb-505124(cas 694433-59-5)、sb-431542(cas 301836-41-9)、sb-202474(emd4 biosciences merck kgaa,darmstadt,germany)、ly-364947(cas 396129-53-6)、in-1130、gw-788388和d4476(emd4 biosciences merck kgaa,darmstadt,germany).
[0129]
本文所述的alk5抑制剂的结构和名称是指连接至抗体和/或接头之前的分子。
[0130]
优选的alk5抑制剂是可以经由游离nh或nh2基团,优选连接至烷基、杂芳基或芳基基团或其一部分的nh或nh2基团(例如,如表2所示的化合物1-23、26-29、31、35、37、39、40、42、43、45-48、50-85、87-90、93、96、98-104、106、108、109、111、112、114、116-120、132、146、149、156、184、187、193、218、260-277、282和283)。可以衍生alk5抑制剂以添加游离nh或nh2基团。衍生化alk5抑制剂的设计应优先考虑抑制剂的构效关系(sar),以降低在添加nh或nh2基团时消除抑制活性的可能性,尽管活性也可以根据经验确定。表1中所示的几种化合物的示例性衍生对应物如下表3所示。
[0131]
val-cit-pab-oh(broadpharm cat.no.bp-23204)、mal-peg4-val-cit-pab-pnp(broadpharm cat.no.bp-23668)、mal-amido-peg2-val-cit-pab-pnp(broadpharm cat.no.bp-23675)、azido-peg3-val-cit-pab-oh(broadpharm cat.no.bp-23206)、azido-peg4-val-cit-pab-oh(broadpharm cat.no.bp-23207)、azido-peg3-val-cit-pab-pnp(broadpharm cat.no.bp-23368)、fmoc-peg4-ala-ala-asn-pab(bp-23328)、azido-peg5-ala-ala-asn-pab(broadpharm cat.no.bp-23329)、fmoc-peg3-ala-ala-asn(trt)-pab(broadpharm cat no.bp-23285)、azido-peg4-ala-ala-asn(trt)-pab(broadpharm cat no.bp-23284)和fmoc-peg3-ala-ala-asn(trt)-pab-pnp(broadpharm cat no.bp-23297)。在一些实施方案中,tdc接头包含peg和肽,例如本节中描述的二肽之一,如val-cit。
[0135]
接头可以包含一种或多种接头组分,如延伸部分和间隔部分。例如,肽基接头可以包含两个或更多个氨基酸的肽基组分和任选的一个或多个延伸部分和/或间隔部分。各种接头组分在本领域中是已知的,其中一些在下文描述。
[0136]
接头可以是“可切割接头”,促进药物在细胞中的释放。例如,可以使用酸不稳定接头(例如腙)、蛋白酶敏感性(例如肽酶敏感性)接头、光不稳定接头、二甲基接头或含二硫化物接头(chari et al.,1992,cancer research 52:127-131;u.s.patent no.5,208,020)。
[0137]
本领域已知的接头和接头组分的实例包括马来酰亚胺基己酰基(mc);马来酰亚胺己酰基-对氨基苄基氨基甲酸酯;马来酰亚胺己酰基-肽-氨基苄基氨基甲酸酯接头,例如马来酰亚胺基己酰基-l-苯丙氨酸-l-赖氨酸-对氨基苄基氨基甲酸酯和马来酰亚胺基己酰基-l-缬氨酸-l-瓜氨酸-对氨基苄基氨基甲酸酯(vc);n-琥珀酰亚胺基3-(2-吡啶基二硫代)丙酸酯(也称为n-琥珀酰亚胺基4-(2-吡啶基二硫代)戊酸酯或spp);4-琥珀酰亚胺基-氧羰基-2-甲基-2-(2-吡啶基二硫代)-甲苯(smpt);n-琥珀酰亚胺基3-(2-吡啶基二硫代)丙酸酯(spdp);n-琥珀酰亚胺基4-(2-吡啶基二硫代)丁酸酯(spdb);2-亚氨基硫烷;s-乙酰琥珀酸酐;二硫化氨基甲酸苄酯;碳酸酯;腙接头;n-(α-马来酰亚胺基乙酰氧基)琥珀酰亚胺酯;n-[4-(对叠氮基水杨酰氨基)丁基]-3'-(2'-吡啶基二硫代)丙酰胺(amas);n[β-马来酰亚胺丙氧基]琥珀酰亚胺酯(bmps);[n-ε-马来酰亚胺基己酰氧基]琥珀酰亚胺酯(emcs);n-[γ-马来酰亚胺丁酰氧基]琥珀酰亚胺酯(gmbs);琥珀酰亚胺基-4-[n-马来酰亚胺甲基]环己烷-1-羧基-[6-氨基己酸酯](lc-smcc);琥珀酰亚胺基6-(3-[2-吡啶基二硫代]-丙酰胺基)己酸酯(lc-spdp);间马来酰亚胺苯甲酰基-n-羟基琥珀酰亚胺酯(mbs);n-琥珀酰亚胺基[4-碘乙酰基]氨基苯甲酸酯(siab);琥珀酰亚胺基4-[n-马来酰亚胺甲基]环己烷-1-羧酸酯(smcc);n-琥珀酰亚胺基3-[2-吡啶基二硫代]-丙酰胺基(spdp);[n-ε-马来酰亚胺基己酰氧基]磺基琥珀酰亚胺酯(sulfo-emcs);n-[γ-马来酰亚胺丁酰氧基]磺基琥珀酰亚胺酯(磺基-gmbs);4-磺基琥珀酰亚胺基-6-甲基-α-(2-吡啶基二硫代)甲苯胺基]己酸酯-)(磺基-lc-smpt);磺基琥珀酰亚胺基6-(3'-[2-吡啶二硫基]-丙酰胺)己酸(sulfo-lc-spdp);间-马来酰亚胺苯甲酰基-n-羟基磺基琥珀酰亚胺酯(sulfo-mbs);n-磺基琥珀酰亚胺基[4-碘乙酰基]氨基苯甲酸酯(磺基-siab);磺基琥珀酰亚胺4-[n-马来酰亚胺甲基]环己烷-1-羧酸酯(sulfo-smcc);磺基琥珀酰亚胺基4-[对马来酰亚胺苯基]丁酸酯(sulfo-smpb);乙二醇-双(琥珀酸n
‑‑
羟基琥珀酰亚胺酯)(egs);酒石酸二琥珀酰亚胺酯(dst);1,4,7,10-四氮杂环十二烷-1,4,7,10-四乙酸(dota);二乙烯三胺-五乙酸(dtpa);硫脲接头;和含有肟的接头。
[0138]
在一些实施方案中,接头在细胞内或细胞外条件下是可切割的,使得接头的切割在合适的环境中从靶向部分释放alk5抑制剂。在其他实施方案中,接头是不可切割的并且释放了药物,例如,通过靶向溶酶体中的部分降解(参见美国专利公开2005/0238649,其通过引用整体并入本文并用于所有目的)。
[0139]
可用于本公开的tdc中的不可切割接头的实例包括n-马来酰亚胺基甲基环己烷1-羧酸酯、马来酰亚胺基己酰基或巯基乙酰胺基己酰基接头。
[0140]
在一些实施方案中,接头通过存在于细胞内环境中(例如,在溶酶体或内体或小窝(caveolea)中)的切割剂是可切割的。接头可以是例如通过细胞内肽酶或蛋白酶切割的肽基接头,其包括但不限于溶酶体或内体蛋白酶。在一些实施方案中,肽基接头包含至少两个氨基酸长或至少三个氨基酸长或更长的肽基组分。
[0141]
切割剂可以包括但不限于组织蛋白酶b和d以及纤溶酶,已知所有这些都水解二肽药物衍生物,导致靶细胞内的活性药物释放(参见,例如,dubowchik and walker,1999,pharm.therapeutics 83:67-123)。例如,肽基接头可通过硫醇依赖性蛋白酶组织蛋白酶-b是可切割的(例如,phe-leu或gly-phe-leu-gly接头)。此类接头的其他实例描述于例如美国专利6,214,345号,其内容通过引用整体并入本文并用于所有目的。
[0142]
在一些实施方案中,可通过细胞内蛋白酶切割的肽基接头是val-cit接头或phe-lys接头(参见,例如,美国专利6,214,345号,其描述了用val-cit接头合成多柔比星)。
[0143]
在其他实施方案中,可切割接头是ph敏感的,即在某些ph值下对水解敏感。通常,ph敏感接头可在酸性条件下水解。例如,可以使用在溶酶体中可水解的酸不稳定接头(例如,腙、氨基脲、氨基硫脲、顺乌头酰胺、原酸酯、缩醛、缩酮等)。(参见,例如,美国专利5,122,368;5,824,805;5,622,929号;dubowchik and walker,1999,pharm.therapeutics 83:67-123;neville et al.,1989,biol.chem.264:14653-14661.)。此类接头在中性ph条件下相对稳定,如在血液中,但在ph低于5.5或5.0(溶酶体的近似ph值)下不稳定。在某些实施方案中,可水解接头是硫醚接头(如,经由酰基腙键连接至治疗剂的硫醚(参见,例如,美国专利5,622,929号)。
[0144]
在其他实施方案中,接头在还原条件下是可切割的(例如,二硫化物接头)。多种二硫化物接头是本领域已知的,其包括,例如,可以使用以下形成的那些:sata(n-琥珀酰亚胺基-5-乙酰硫代乙酸酯)、spdp(n-琥珀酰亚胺基-3-(2-吡啶基二硫代)丙酸酯)、spdb(n-琥珀酰亚胺基-3-(2-吡啶基二硫代)丁酸酯)和smpt(n-琥珀酰亚胺基-氧羰基-α-甲基-α-(2-吡啶基-二硫代)甲苯)-、spdb和smpt。(参见,例如,thorpe et al.,1987,cancer res.47:5924-5931;wawrzynczak et al.,in immunoconjugates:antibody conjugates in radioimagery and therapy of cancer(c.w.vogel ed.,oxford u.press,1987。还参见例如美国专利4,880,935号。)
[0145]
在其他实施方案中,接头是丙二酸酯接头(johnson et al.,1995,anticancer res.15:1387-93)、马来酰亚胺苯甲酰基接头(lau et al.,1995,bioorg-med-chem.3(10):1299-1304),或3'-n-酰胺类似物(lau et al.,1995,bioorg-med-chem.3(10):1305-12)。
[0146]
在一些实施方案中,接头是可用于将许多药物分子连接到单个靶向部分分子(例如,单个抗体分子)的多价接头。例如,由mersana开发的fleximer接头技术基于经由一系列酯键将药物分子结合到增溶聚缩醛主链中。该方法能够实现高负载的tdc(例如,具有高达
20的药物抗体比率(dar)),同时保持良好的物理化学特性。示例性多价接头描述于例如wo 2009/073445;wo 2010/068795;wo 2010/138719;wo 2011/120053;wo 2011/171020;wo 2013/096901;wo 2014/008375;wo 2014/093379;wo 2014/093394;和wo 2014/093640,其内容通过引用整体并入本文。
[0147]
通常,接头对细胞外环境基本上不敏感。如本文所用,“对细胞外环境基本不敏感”在接头的上下文中是指,当在细胞外环境(例如,在血浆中)中存在tdc时,在tdc的样品中切割不超过约20%、15%、10%、5%、3%或不超过约1%的接头。
[0148]
可以确定接头是否对细胞外环境基本上不敏感,例如,通过将tdc与血浆一起温育预定时间段(例如,2、4、8、16或24小时),然后量化存在于血浆中的游离药物的量。
[0149]
在其他非相互排斥的实施方案中,接头可以促进细胞内化。在某些实施方案中,接头在与治疗剂缀合时促进细胞内化(即,在如本文所述的tdc的接头-治疗剂部分的环境中)。在其他实施方案中,接头在与alk5抑制剂和抗体两者缀合时促进细胞内化。
[0150]
在许多实施方案中,接头是自我牺牲的。如本文所用,术语“自我牺牲”是指能够将两个隔开的化学部分共价连接在稳定的三联分子的双功能化学部分。如果切割它与第一部分的键,它将自发地与第二化学部分分离。例如,参见pct公开wo 2007/059404、wo 2006/110476、wo 2005/112919、wo 2010/062171、wo 2009/017394、wo 2007/089149、wo 2007/018431、wo 2004/043493和wo 2002/083180号,其针对药物可切割底物缀合物,其中药物和可切割底物任选地通过自我牺牲接头连接并且均通过引用明确并入。可用于生成自我牺牲接头的自我牺牲间隔单元的实例在下面的式i中描述。
[0151]
可与本组合物和方法使用的多种示例性接头描述于pct公开wo 2004/010957号、美国专利公开us 2006/0074008号、美国专利公开号us 2005/0238649号和美国专利公开us 2006/0024317号(每一个都通过引用整体并入本文,并用于所有目的)。
[0152]
本公开的tdc可以具有以下式i,其中抗体或其他靶向部分(在式i中显示为“ab”)通过任选的接头(l)与一种或多种alk5抑制剂药物部分(d)缀合
[0153]
ab-(l-d)
p
ꢀꢀꢀꢀi[0154]
因此,靶向部分可以直接或经由接头与药物缀合。在式i中,p是每个靶向部分的药物(即,alk5抑制剂)部分的平均数,其范围可以是例如每个靶向部分约1至约20个药物部分,并且在某些实施方案中,每个靶向部分2至约8个药物部分。载药量的进一步细节在下面的第5.5节中描述。
[0155]
在一些实施方案中,接头组分可包含将靶向部分(例如,经由半胱氨酸残基)连接至另一接头组分或药物部分的“延伸段(stretcher)”。示例性的延伸段如下所示(其中左侧波浪线表示与靶向部分共价连接的位点,而右侧波浪线表示与另一个接头组分或药物部分共价连接的位点):
[0156][0157][0158]
参见,美国专利9,109,035号;ducry et al.,2010,bioconjugate chem.21:5-13。
[0159]
在一些实施方案中,接头组分可以包含氨基酸单元。在一个此类实施方案中,氨基酸单元允许通过蛋白酶切割接头,从而在暴露于细胞内蛋白酶,如溶酶体酶时促进药物从tdc释放。参见,例如,doronina et al.,2003,nat.biotechnol.21:778-784。示例性氨基酸单元包括但不限于二肽、三肽、四肽和五肽。示例性二肽包括:缬氨酸-瓜氨酸(vc或val-cit)、丙氨酸-苯丙氨酸(af或ala-phe);苯丙氨酸-赖氨酸(fk或phe-lys);或n-甲基-缬氨酸-瓜氨酸(me-val-cit)。示例性三肽包括:甘氨酸-缬氨酸-瓜氨酸(gly-val-cit)和甘氨酸-甘氨酸-甘氨酸(gly-gly-gly)。示例性四肽包括甘氨酸-甘氨酸-苯丙氨酸-甘氨酸(gly-gly-phe-gly)。氨基酸单元可以包含天然存在的氨基酸残基,以及少量氨基酸和非天然存在的氨基酸类似物,如可以设计和优化瓜氨酸氨基酸单元对特定酶(例如,组织蛋白酶b、c和d,或纤溶酶蛋白酶)的酶促切割的选择性。
[0160]
在一些实施方案中,接头组分可以包含将靶向部分与药物部分直接或通过延伸和/或氨基酸单元连接的“间隔”单元。间隔单元可以是“自我牺牲”或“非自我牺牲”。“非自我牺牲”间隔单元是如下的间隔单元,其中部分或全部间隔单元在tdc的酶促(例如,蛋白水解)切割时保持与药物部分结合。非自我牺牲间隔单元的实例包括但不限于甘氨酸间隔单元和甘氨酸-甘氨酸间隔单元。“自我牺牲”间隔单元允许药物部分的释放,而无需单独的水
解步骤。在某些实施方案中,接头的间隔单元包含对氨基苄基单元。在一个此类实施方案中,对氨基苯甲醇经由酰胺键连接到氨基酸单元,并且在苯甲醇和细胞毒剂之间制成氨基甲酸酯、氨基甲酸甲酯或碳酸酯。参见,例如,hamann et al.,2005,expert opin.ther.patents 15:1087-1103。在一个实施方案中,间隔单元是对氨基苄氧羰基(pab)。在某些实施方案中,用qm取代对氨基苄基单元的亚苯基部分,其中q是-c
1-c8烷基、
‑‑o‑‑
(c1-c8烷基)、-卤素、-硝基或-氰基;m为0-4范围的整数。自我牺牲间隔单元的实例进一步包括但不限于在电子上与对氨基苯甲醇相似的芳族化合物(参见,例如美国专利公开号us 2005/0256030号),如2-氨基咪唑-5-甲醇衍生物(hay et al.,1999,bioorg.med.chem.lett.9:2237)和邻-或对-氨基苄缩醛。可以使用在酰胺键水解时发生环化的间隔,如取代和未取代的4-氨基丁酸酰胺(rodrigues et al.,1995,chemistry biology 2:223);适当取代的双环[2.2.1]和双环[2.2.2]环系统(storm et al.,1972,amer.chem.soc.94:5815);和2-氨基苯基丙酸酰胺(amsberry et al.,1990,j.org.chem.55:5867)。消除在甘氨酸的α位取代的含胺药物(kingsbury et al.,1984,j.med.chem.27:1447)也是可用于tdc的自我牺牲间隔的实例。
[0161]
在一个实施方案中,间隔单元是如下所述的支链双(羟甲基)苯乙烯(bhms)单元,其可用于掺入和释放多种药物。
[0162][0163]
其中ab和d如上式i定义;a为延伸段,a为0至1的整数;w为氨基酸单元,w为0至12的整数;q是-c
1-c8烷基、-o
‑‑
(-c
1-c8烷基)、-卤素、-硝基或-氰基;m为0-4的整数;n为0或1;p范围为从1至约20。
[0164]
接头可包含任何一种或多种上述接头组分。在某些实施方案中,接头如以下tdc式中的括号中所示:
[0165]
ab
–
(
–
[aa-ww-yy]-d)
p
ꢀꢀꢀꢀ
ii
[0166]
其中ab、a、a、w、w、d和p如前一段所定义;y为间隔单元,y为0、1或2;此类接头的示例性实施方案在美国专利公开2005/0238649a1号中描述,其通过引用并入本文。
[0167]
示例性接头组分及其组合在式ii的tdc的上下文中如下所示:
[0168][0169]
接头组分,包括延伸段、间隔段和氨基酸单元,可以通过本领域已知的方法合成,如在美国专利公开2005/0238649号中描述的那些。
[0170]
5.5.载药量
[0171]
载药量由p表示并且是分子中每个靶向部分(例如,每个抗体)的alk5抑制剂部分的平均数。载药量(“p”)可以是每个靶向部分有1、2、3、4、5、6、7、8、9、10、11、12、13、14、15、16、17、18、19、20或更多个部分(d),尽管通常平均数是分数或小数。通常,alk5抑制剂负载平均每个靶向部分2至8个药物部分,更优选每个抗体2至4个药物部分或每个靶向部分5至7
个药物部分。
[0172]
如本领域技术人员将理解的,在许多情况下,提及tdc是tdc分子群或集合的简写(有时在药物组合物的上下文中),每个分子由共价连接到一个或多个alk5抑制剂部分的靶向部分组成,载药率代表群或集合中的平均载药量,尽管基于个体分子的比可能随着群中的一个tdc分子到另一个分子而不同。在一些实施方案中,该群或集合含有tdc分子,该tdc分子包含共价连接至1至30个药物部分之间的任意位置的抗体,在一些实施方案中,1至20个、1至15个、2至12个、2至8个、4至15个或6至12个药物部分之间的任意位置的抗体。优选地,群中的平均值如前一段所述,例如每个靶向部分2至8个药物部分,更优选每个靶向部分4至8个药物部分或每个靶向部分5至7个药物部分。
[0173]
一些tdc群可以是包含如本文所述的tdc和缺乏药物部分的靶向部分分子的组合物的形式,例如,alk5抑制剂连接不成功的抗体。
[0174]
来自缀合反应的tdc制剂中每个靶向部分的alk5抑制剂部分的平均数可以通过常规方法,如质谱、疏水作用色谱(hic)和elisa测定表征。
[0175]
也可以确定tdc在p方面的定量分布。在一些情况下,可通过手段,如电泳,实现从有其他alk5抑制剂负载的tdc中分离、纯化和表征其中p为特定值的均质tdc。
[0176]
对于一些药物缀合物,p可能受靶向部分上连接位点的数量限制。例如,在连接物是半胱氨酸硫醇的情况下,如在上述示例性实施方案中,靶向部分(例如,抗体)可以仅具有一个或几个半胱氨酸硫醇基团,或者可以仅具有一个或几个充分反应性的硫醇基团,通过上述基团可以连接接头。在某些实施方案中,较高的载药量,例如p>5,可能导致某些药物缀合物的聚集、不溶解、毒性或细胞渗透性丧失。在某些实施方案中,本公开的tdc的载药量为1至约8;约2至约6;约3至约5;约3至约4;约3.1至约3.9;约3.2至约3.8;约3.2至约3.7;约3.2至约3.6;约3.3至约3.8;或约3.3至约3.7的范围内。事实上,已经显示对于某些tdc,每个抗体的药物部分的最佳比可能小于8,并且可能为约2至约5。参见美国专利公开us 2005/0238649号(通过引用整体并入本文)。
[0177]
在某些实施方案中,在缀合反应期间,少于理论最大值的药物部分与靶向部分缀合。如下所述,靶向部分可以含有,例如不与药物-接头中间物或接头试剂反应的赖氨酸残基。通常,抗体不含有许多可能与药物部分连接的游离和反应性半胱氨酸硫醇基团;事实上,抗体中的大多数半胱氨酸硫醇残基以二硫键的形式存在。在某些实施方案中,可以在部分或全部还原条件下用还原剂,如二硫苏糖醇(dtt)或三羰基乙基膦(tcep)还原抗体或其他靶向部分,以生成反应性半胱氨酸硫醇基团。在某些实施方案中,抗体或其他靶向部分经受变性条件以显示反应性亲核基团,如赖氨酸或半胱氨酸。
[0178]
可以以不同方式控制tdc的负载(药物/抗体比),例如,通过:(i)限制药物-接头中间物或接头试剂相对于靶向部分的摩尔过量,(ii)限制缀合反应时间或温度,(iii)半胱氨酸硫醇修饰的部分或限制性还原条件,(iv)通过重组技术工程化靶向部分的氨基酸序列,从而修饰半胱氨酸残基的数量和位置以控制数量和/或接头-药物连接物的位置(如,如pct公开wo 2006/034488号中所公开制备的thiomab或thiofab(通过引用整体并入本文))。
[0179]
应当理解,当多于一个亲核基团与药物-接头中间物或接头试剂反应,然后与药物部分试剂反应时,所得产物是tdc化合物的混合物,其中一个或多个药物部分与靶向部分连接分布。每个靶向部分的平均药物数量可以通过双重elisa抗体测定从混合物中计算出来,
该测定对靶向部分具有特异性并且对药物具有特异性。可以通过质谱识别混合物中的个体tdc分子并通过hplc分离,例如疏水作用色谱。
[0180]
在一些实施方案中,可通过电泳或色谱法从缀合混合物中分离有单一加载值的均质tdc。
[0181]
5.6.制剂和施用
[0182]
tdc的合适施用途径包括但不限于口服、肠胃外、直肠、经粘膜、肠施用、髓内、鞘内、直接心室内、静脉内、玻璃体内、腔内、腹膜内或瘤内注射。优选的施用途径是肠胃外的,更优选静脉内的。或者,可以以局部而非全身方式施用化合物,例如经由将化合物直接注射到受纤维化影响的区域或经由将化合物直接注射到实体瘤中。
[0183]
可以根据已知方法配制免疫缀合物以制备药学上有用的组合物,由此在混合物中将tdc与药学上合适的赋形剂组合成。无菌磷酸盐缓冲盐水是药学上合适的赋形剂的一个实例。其他合适的赋形剂为本领域技术人员所熟知。例如,参见ansel et al.,pharmaceutical dosage forms and drug delivery systems,5th edition(lea&febiger 1990)和gennaro(ed.),remington's pharmaceutical sciences,18th edition(mack publishing company 1990)以及其修订版。
[0184]
在优选的实施方案中,使用缓冲液在good氏生物缓冲液(ph 6-7)中配制tdc,其中缓冲液选自由n-(2-乙酰氨基)-2-氨基乙磺酸(aces);n-(2-乙酰氨基)亚氨基二乙酸(ada);n,n-双(2-羟乙基)-2-氨基乙磺酸(bes);4-(2-羟乙基)哌嗪-1-乙磺酸(hepes);2-(n-吗啉代)乙磺酸(mes);3-(n-吗啉代)丙磺酸(mops);3-(n-吗啉基)-2-羟基丙磺酸(mopso);和哌嗪-n,n'-双(2-乙磺酸)[pipes]组成的组。更优选的缓冲液是mes或mops,优选浓度范围为20至100mm,更优选约25mm。最优选的是25mm mes,ph 6.5。该制剂可进一步包含25mm海藻糖和0.01%v/v聚山梨醇酯80作为赋形剂,由于添加了赋形剂,最终修改缓冲液浓度为22.25mm。优选的储存方法是作为缀合物的冻干制剂,储存在-20℃至2℃的温度范围内,最优选储存在2℃至8℃。
[0185]
可以配制tdc用于经由,例如推注、缓慢输注或连续输注进行静脉内施用。优选地,在少于约4小时的时间段内输注tdc,更优选地,在少于约3小时的时间段内输注。例如,前25-50mg可以在30分钟内输注,最好甚至15min,其余的在接下来的2-3小时内输注。用于注射的制剂可以以单位剂型存在,例如,在安瓿中或在多剂量容器中,并添加有防腐剂。所述组合物可以采取如在油性或水性媒介物中的悬浮液、溶液或乳液的形式,并且可以含有配方剂,如悬浮剂、稳定剂和/或分散剂。或者,活性成分可以是粉末形式,用于在使用前与合适的媒介物,例如无菌无热原水混合。
[0186]
可以采用额外药物方法来控制tdc的作用持续时间。控释制剂可以通过使用聚合物复合或吸附tdc来制备。例如,生物相容性聚合物包括聚(乙烯-共-乙酸乙烯酯)基质和硬脂酸二聚体和癸二酸的聚酐共聚物基质。sherwood et al.,1992,bio/technology 10:1446。tdc从此类基质中释放的速率取决于tdc的分子量、基质中tdc的量和分散颗粒的尺寸。saltzman et al.,1989,biophys.j.55:163;sherwood et al.,同上。ansel et al.,pharmaceutical dosage forms and drug delivery systems,5th edition(lea&febiger 1990)和gennaro(ed.),remington's pharmaceutical sciences,18th edition(mack publishing company 1990)及其修订版中描述了其他固体剂型。
[0187]
一般而言,对人施用的tdc的剂量将根据此类因素,如患者的年龄、体重、身高、性别、一般健康状况和既往病史而变化。可能需要为接受者提供约0.3mg/kg至5mg/kg范围内的tdc剂量作为单次静脉内输注,尽管根据情况也可以施用更低或更高的剂量。例如,对于70kg的患者,根据0.3-5mg/kg的剂量是21-350mg,或者对于1.7-m的患者,根据12-206mg/m2的剂量是21-350mg。可以根据需要重复剂量,例如每周一次持续2-10周,每周一次持续8周,或每周一次持续4周。根据维持疗法的需要,也可以不那么频繁地给予,如每隔一周持续几个月,或每月或每季度持续几个月。优选的剂量可以包括但不限于0.3mg/kg、0.5mg/kg、0.7mg/kg、1.0mg/kg、1.2mg/kg、1.5mg/kg、2.0mg/kg、2.5mg/kg、3.0mg/kg、3.5mg/kg、4.0mg/kg、4.5mg/kg和5.0mg/kg。更优选的剂量是每周施用0.6mg/kg和以1.2mg/kg不太频繁给药。可以使用0.3至5mg/kg范围内的任何量。该剂量优选每周一次多次施用。可以根据4周、更优选8周、更优选16周或更长时间的时间表施用最小剂量,施用频率取决于毒副作用和从中恢复的情况,主要与血液学毒性有关。施用时间表可以包括每周施用一次或两次,例如在选自由以下组成的组的周期:(i)每周;(ii)每隔一周;(iii)治疗一周,然后休息两、三或四周;(iv)治疗两周,然后休息一、二、三或四周;(v)治疗三周,然后休息一、二、三、四或五周;(vi)治疗四周,然后休息一、二、三、四或五周;(vii)治疗五周,然后休息一、二、三、四或五周;(viii)每月一次。该周期可以重复2、4、6、8、10或12或更多次。
[0188]
或者,tdc可以每2或3周作为一个剂量施用,重复总共至少3个剂量。或者,每周两次持续4-6周。该剂量可以每隔一周施用一次,甚至可以不那么频繁地施用,因此患者可以从任何与药物相关的毒性中恢复过来。或者,可以减少剂量时间表,即每2或3周一次持续2-3个月。给药方案可以任选地以其他间隔重复,并且可以通过各种肠胃外途径给予剂量,并适当调整剂量和时间表。
[0189]
5.7.治疗方法
[0190]
5.7.1.纤维化
[0191]
本公开的tdc可用于治疗各种纤维化病症,例如与系统性硬化症(也称为硬皮病)或nash相关的纤维化。患有系统性硬化症的患者经常承受肺纤维化、皮肤纤维化和食道纤维化之苦,尽管纤维化几乎可以发生在任何器官中。nash患者经常遭受肝纤维化之苦。tdc可用作单一疗法或作为组合疗法方案的一部分,例如与标准护理剂或方案一起使用。在一些实施方案中,组合疗法包括tdc与吡非尼酮、尼达尼布、pentraxin-2、pamrevlumab、泼尼松、可的松、环磷酰胺、硫唑嘌呤或其组合组合施用。在一些实施方案中,组合疗法包括tdc与吡非尼酮和/或尼达尼布组合施用。
[0192]
可以使用本公开的tdc治疗的病况的实例包括但不限于肺纤维化(例如ipf)、肝纤维化(例如与nash相关的)、肾纤维化、心脏纤维化、皮肤纤维化、食道纤维化和全身硬化。本公开的tdc可以在纤维化的体征和/或症状发展之前施用于患有,例如诊断患有与纤维化相关的疾病(例如系统性硬化症或nash)的受试者。或者或除此之外,在观察到纤维化的体征和/或症状后,可以将tdc施用于患有,例如诊断患有与纤维化相关的疾病的受试者。
[0193]
本公开的tdc与一种或多种疗法组合的使用不限制其中施用疗法的顺序。例如,本公开的tdc可以在用一种或多种疗法治疗受试者之前、期间或之后施用。在一些实施方案中,在用另一种疗法(例如,如上所述的第二治疗剂)治疗患者之前(例如,5分钟、15分钟、30分钟、45分钟、1小时、2小时、4小时、6小时、12小时、24小时、48小时、72小时、96小时、1周、2
周、3周、4周、5周、6周、8周或12周前)、同时或之后(例如,5分钟、15分钟、30分钟、45分钟、1小时、2小时、4小时、6小时、12小时、24小时、48小时、72小时、96小时、1周、2周、3周、4周、5周、6周、8周或12周后)施用本公开的tdc。在一些实施方案中,本公开的tdc并入与第二治疗剂相同的方案中。
[0194]
5.7.2.癌症
[0195]
本公开的tdc(例如,靶向fap的tdc)可用于治疗各种癌症。tdc可用作单一疗法或作为组合疗法方案的一部分,例如与标准护理剂或方案一起使用。在一些实施方案中,组合疗法包括tdc与免疫疗法(例如检查点抑制剂疗法)、嵌合抗原受体(car)疗法、过继性t细胞疗法(例如,自体t细胞疗法)、溶瘤病毒疗法、树突细胞疫苗疗法、干扰素基因刺激剂(sting)激动剂疗法、toll样受体(tlr)激动剂疗法、瘤内cpg疗法、细胞因子疗法(例如,il2、il12、ifn-α或inf-γ疗法)或其组合联合施用。在一些实施方案中,组合疗法包括tdc与具有细胞毒性有效载荷的adc,例如,靶向fap的adc(如,omtx705(oncomatryx))组合施用。在一些实施方案中,组合疗法包括tdc与免疫保护化疗(例如,抗代谢物,(如5-氟尿嘧啶、吉西他滨或甲氨蝶呤)、烷化剂(如环磷酰胺、达卡巴嗪、氮芥、地吖醌或替莫唑胺)、蒽环类药物(如多柔比星或表柔比星)、抗微管剂(如长春碱)、铂化合物(如顺铂或奥沙利铂)、紫杉烷(如紫杉醇或多西紫杉醇)或拓扑异构酶抑制剂(如依托泊苷或米托蒽醌)或长春花生物碱(如长春新碱)组合施用。
[0196]
可以使用本公开的tdc治疗的癌症的实例包括但不限于尿路上皮癌(例如,膀胱癌、尿道癌和输尿管癌)、肺癌(例如,非小细胞肺癌(nsclc)(如腺癌、鳞状细胞癌、大细胞癌)和小细胞肺癌)、乳腺癌、结直肠癌(例如,腺癌、类癌瘤、胃肠道间质瘤和结直肠淋巴瘤)、胰腺癌、前列腺癌和食道癌。可以用本公开的tdc治疗的癌症的其他实例包括头颈癌、卵巢癌、肾脏癌和胃腺癌。
[0197]
本公开的tdc可以与检查点抑制剂,例如靶向pd1、pdl1、ctla4、tigit、lag3、ox40、cd40或vista的检查点抑制剂组合使用。检查点抑制剂包括抗体和小分子。靶向pd1的示例性检查点抑制剂包括派姆单抗(pembrolizumab)、纳武单抗(nivolumab)、西米普利单抗(cemiplimab)和多塔利单抗(dostarlimab)。靶向pdl1的示例性检查点抑制剂包括阿特珠单抗(atezolizumab)、阿维单抗(avelumab)、德瓦鲁单抗(durvalumab)、bms-1001和bms-1166。靶向ctla4的示例性检查点抑制剂是伊匹单抗(ipilimumab)。靶向tigit的示例性检查点抑制剂包括etigilimab、tiragolumab和ab154。靶向lag3的示例性检查点抑制剂包括lag525、sym022、relatlimab和tsr-033。靶向ox40的示例性检查点抑制剂包括medi6469、pf-04518600和bms986178。靶向cd40的示例性检查点抑制剂包括selicrelumab、cp-870,893和apx005m。靶向vista的示例性检查点抑制剂是hmbd-002。对于尿路上皮癌的治疗,本公开的tdc可以与标准护理治疗组合使用,包括但不限于顺铂、丝裂霉素-c、卡铂(carboplatin)、多西紫杉醇、紫杉醇、多柔比星、5-fu、甲氨蝶呤、长春碱、异环磷酰胺(ifosfamide)和培美曲塞(pemetrexed)。此外,tdc可与检查点抑制剂(如伊匹单抗)组合使用。
[0198]
对于非小细胞肺癌(nsclc)的治疗,本公开的tdc可与标准护理化疗治疗组合使用,标准护理化疗治疗包括如顺铂、卡铂、紫杉醇、吉西他滨、长春瑞滨(vinorelbin)、伊立替康(irinotecan)、依托泊苷或长春碱。此外,tdc可以与靶向疗法,如贝伐珠单抗
(bevacizumab)或爱必妥(erbitux)组合使用。此外,tdc可与检查点抑制剂,如派姆单抗、纳武单抗、西米普利单抗、多塔利单抗、阿特珠单抗、阿维单抗、德瓦鲁单抗或伊匹单抗组合使用。
[0199]
对于乳腺癌的治疗,本公开的tdc可以与标准护理化疗剂,如蒽环类(多柔比星或表柔比星)和紫杉烷(紫杉醇或多西紫杉醇)以及氟尿嘧啶、环磷酰胺和卡铂组合使用。此外,本公开的tdc可以与靶向疗法组合使用。her2/neu阳性肿瘤的靶向疗法包括曲妥珠单抗(trastuzumab)和帕妥珠单抗(pertuzumab),雌激素受体(er)阳性肿瘤的靶向疗法包括他莫昔芬(tamoxifen)、托瑞米芬(toremifene)和氟维司群(fulvestrant)。此外,tdc可以与检查点抑制剂,如阿特珠单抗组合使用。
[0200]
对于结直肠癌的治疗,本公开的tdc可以与标准护理治疗组合使用,标准护理治疗包括但不限于5-fu、卡培他滨(capecitabine)、伊立替康、奥沙利铂、曲氟尿苷(trifluridine)和替吡嘧啶(tipiracil)。此外,本公开的tdc可以与靶向疗法组合使用。靶向治疗包括贝伐珠单抗、ramucirumab和ziv-阿柏西普(ziv-aflibercept)。此外,tdc可与检查点抑制剂,如派姆单抗、纳武单抗或伊匹单抗组合使用。
[0201]
对于胰腺癌,本公开的tdc可以与标准护理化疗剂,如吉西他滨、5-氟尿嘧啶、伊立替康、奥沙利铂、紫杉醇、卡培他滨、顺铂或多西紫杉醇组合使用。此外,tdc可以与靶向疗法组合使用,如抑制egfr的厄洛替尼(erlotinib)。
[0202]
对于前列腺癌,本公开的tdc可以与标准护理化疗剂,包括多西紫杉醇组合使用,任选地与类固醇泼尼松或卡巴他赛(cabazitaxel)组合使用。此外,tdc可与检查点抑制剂,如伊匹单抗组合使用。
[0203]
对于食管癌,本公开的tdc可以与标准护理化疗剂,如卡铂和紫杉醇,顺铂和5-fu,表柔比星、顺铂和5-fu,多西紫杉醇、顺铂和5-fu,顺铂与卡培他滨,奥沙利铂和5-fu或卡培他滨,伊立替康或曲氟尿苷和替吡嘧啶组合使用。此外,tdc可与靶向疗法,如曲妥珠单抗或ramucirumab组合使用。此外,tdc可与检查点抑制剂,如派姆单抗组合使用。
[0204]
本公开的tdc与一种或多种疗法的组合使用不限制施用疗法的顺序。例如,本公开的tdc可以在用一种或多种疗法治疗受试者之前、期间或之后施用。在一些实施方案中,在用另一种疗法(例如,如上所述的第二治疗剂)治疗患者之前(例如,5分钟、15分钟、30分钟、45分钟、1小时、2小时、4小时、6小时、12小时、24小时、48小时、72小时、96小时、1周、2周、3周、4周、5周、6周、8周或12周前)、同时或之后(例如,5分钟、15分钟、30分钟、45分钟、1小时、2小时、4小时、6小时、12小时、24小时、48小时、72小时、96小时、1周、2周、3周、4周、5周、6周、8周或12周后)施用本公开的tdc。在一些实施方案中,本公开的tdc并入与第二治疗剂相同的方案中
6.实施例
[0205]
在整个实施例部分中可以找到以下缩写:
[0206]
boc—叔丁氧羰基
[0207]
dcm—二氯甲烷
[0208]
dma—二甲胺
[0209]
dmf—二甲基甲酰胺
[0210]
dipea—n,n-二异丙基乙胺
[0211]
etoac—乙酸乙酯
[0212]
etoh—乙醇
[0213]
fmoc—芴基甲氧羰基
[0214]
hobt—羟基苯并三唑
[0215]
meoh—甲醇
[0216]
nahmds—六甲基二硅氮化钠(sodium hexamethyldisilazide)
[0217]
rt—室温,大约21℃
[0218]
tbtu—o-(苯并三唑-1-基)-n,n,n',n'-四甲基脲四氟硼酸盐(o-(benzotriazol-1-yl)-n,n,n
′
,n
′‑
tetramethyluronium tetrafluoroborate)
[0219]
tea—三乙胺
[0220]
thf—四氢呋喃
[0221]
tfa—三氟乙酸
[0222]
tms-咪唑-1-(三甲基甲硅烷基)咪唑
[0223]
6.1.实施例1:4-(6-甲基吡啶-2-基)-5-(1,5-萘啶-2-基)-1,3-噻唑-2-胺(化合物a)的合成
[0224]
化合物a根据以下方案1中的通用方法制备:
[0225][0226]
6.1.1.2-甲基-1,5-萘啶(a1)
[0227]
在室温下搅拌浓硫酸(2.5ml)、间硝基苯磺酸钠(sodiumm-nitrobenzenesulfonate)(2.08g,9.24mmol)、硼酸(445mg,7.21mmol)和七水合硫酸亚铁(167mg,0.60mmol)的混合物。向反应混合物中加入甘油(1.5ml),然后加入5-氨基-2-甲基吡啶(a-sm)(500mg,4.62mmol)和水(2.5ml)并在135℃加热18h。如通过tlc测量的,在反应完成后,将反应混合物冷却至大约21℃,使用4n naoh碱化并用etoac(2x100ml)萃取。合并有机萃取物,用水(200ml)洗涤,经na2so4干燥并减压蒸发,得到粗化合物a1。粗品通过硅胶柱色谱法使用(2%meoh/ch2cl2)纯化,得到化合物a1,其为淡棕色结晶固体(200mg,30%)。
[0228]1h nmr(500mhz,cdcl3):δ8.92(d,j=3.0hz,1h),8.35(d,j=9.0hz,1h),8.31(d,j=5.9hz,1h),7.62(dd,j=8.5,4.5hz,1h),7.54(d,j=5.9hz,1h),2.8(s,3h)
[0229]
lc-ms(esi):m/z 145[m h]
[0230]
6.1.2.1-(6-甲基吡啶-2-基)-2-(1,5-萘啶-2-基)乙烷-1-酮(a2)
[0231]
将a1(200mg,1.38mmol)和6-甲基吡啶甲酸甲酯(methyl6-methylpicolinate)(209mg,1.38mmol)在无水thf(10ml)中的溶液置于n2气氛下并冷却至-78℃。双(三甲基甲硅烷基)酰胺钾(在甲苯中0.5m,6.9ml,3.47mmol)在5分钟的时间段内逐滴加入。将反应混合物在-78℃搅拌1h,然后升温至大约21℃并保持20h。反应完成后(如通过tlc测量),反应混合物用饱和氯化铵溶液(20ml)淬灭。水层用etoac(2
×
20ml)萃取。合并的有机萃取物用水(100ml)洗涤,经na2so4干燥并蒸发,得到粗化合物a2。粗材料通过柱色谱法(1%meoh/ch2cl2)纯化,得到化合物a2,其为橙黄色固体(110mg,30.5%)。
[0232]1h nmr(400mhz,cdcl3:enol形式):δ15.74(brs,-oh),8.69(t,j=3.6,1h),8.12(d,j=9.2hz,1h),8.06(dd,j=8.4,4.4hz,2h),7.82(t,j=7.6hz,1h),7.55(dd,j=8.4,4.8hz,1h)7.45(d,j=9.6hz,1h),7.3(dd,j=7.6,4.0hz,1h),7.16(s,1h),2.75(s,3h)
[0233]
lc-ms(esi):m/z 264[m h]
[0234]
6.1.3.4-(6-甲基吡啶-2-基)-5-(1,5-萘啶-2-基)-1,3-噻唑-2-胺(化合物a)
[0235]
用溴(0.025ml,0.501mmol)处理a2(110mg,0.418mmol)在1,4-二恶烷(10ml)中的溶液。将所得反应混合物在约21℃搅拌1h,然后减压浓缩,得到粗a3(120mg),其无需进一步纯化即可用于下一步。将粗a3(120mg)溶解在乙醇(15ml)中。然后加入硫脲(3.5mg,0.046mmol)并将反应混合物在78℃加热4h(直到通过tlc观察到起始材料完全消耗)。将反应混合物冷却至大约21℃并在温和搅拌下加入氨溶液(25%,1.5ml)。蒸发溶剂,然后将残余物溶解在ch2cl2(2x20ml)中并用水(50.0ml)洗涤。然后用1n hcl(30mlx2)洗涤分离的有机层。合并的水层用35%水性(aq.)氢氧化钠(20ml)碱化并用ch2cl2(2x20ml)萃取。有机层用硫酸钠干燥并蒸发,得到粗化合物a。将粗化合物a从乙腈(2ml)中重结晶,得到纯化的化合物a,其为黄色结晶固体(经2步的35mg,产率49%)。
[0236]1h nmr(400mhz,cdcl3):δ8.86(dd,j=4.4,1.6hz,1h),8.29(t,j=8.4hz,1h),8.06(d,j=9.2hz,1h),7.64(t,j=7.6hz,1h),7.60-7.55(m,2h),7.46(d,j=8hz,1h),7.20(d,j=7.6,1h),5.32(brs,2h),2.57(s,3h)
[0237]
lc-ms(esi):m/z 320[m h]
[0238]
uplc纯度:97.6%
[0239]
6.2.实施例2:合成n-甲基-2-(4-{4-[3-(吡啶-2-基)-1h-吡唑-4-基]吡啶-2-基}苯氧基)乙烷-1-胺(化合物b)
[0240]
化合物b根据以下方案2中的一般方法制备:
[0241][0242]
6.2.1.(2-氯乙基)(甲基)氨基甲酸叔丁酯(b7)
[0243]
向boc-酸酐(1.7ml,7.30mmol)在thf(4ml)中的搅拌溶液中同时加入b6(1g,7.69mmol)在水(4ml)中的溶液和tea(1ml,7.69mmol)在thf(4ml)中的溶液,历时1小时。将所得混合物在大约21℃搅拌16小时。反应混合物用饱和nacl溶液(20ml)稀释并用dcm(3
×
50ml)萃取。合并的有机萃取物经na2so4干燥,真空浓缩,得到粗化合物,将其通过硅胶柱色谱法使用10%etoac/己烷纯化,得到化合物b7,其为淡黄色液体(1g,5.18mmol,71%)。
[0244]1h nmr(400mhz,cdcl3):δ3.58-3.52(m,4h),2.93(s,3h),1.46(s,9h)
[0245]
6.2.2.甲基(2-(4-(4,4,5,5-四甲基-1,3,2-二恶硼烷-2-基)苯氧基)乙基)氨基甲酸叔丁基酯(int-b)
[0246]
在氩气气氛下向4-羟基苯基硼酸频哪醇酯(789mg,3.58mmol)在dmf(13ml)中的搅拌溶液中加入b7(900mg,4.66mmol)、ki(18mg,0.10mmol)和cs2co3(2.57g,7.88mmol)。将反应混合物加热至65℃并搅拌16小时。将反应混合物倒入水(20ml)中并用etoac(3
×
20ml)萃取。将合并的有机层在减压下浓缩以获得粗品,将其通过柱色谱法使用7%etoac/己烷纯化,得到int-b,其为淡黄色固体(580mg,1.53mmol,43%)。
[0247]1h nmr(400mhz,cdcl3):δ7.74(d,j=8.4hz,2h),6.87(d,j=8.8hz,2h),4.16-4.06(m,2h),3.65-3.59(m,2h),2.97(s,3h),1.45(s,9h),1.33(s,12h)
[0248]
6.2.3.2-(2-溴吡啶-4-基)-1-(吡啶-2-基)乙烷-1-酮(b2)
[0249]
在-78℃在氩气下向2-溴-4-甲基吡啶(b1)(2g,11.62mmol)在thf(30ml)中搅拌的溶液中逐滴加入nahmds(在thf中2m,12.7ml,25.58mmol)的溶液。将黄色溶液在-78℃搅拌30分钟。然后加入吡啶甲酸乙酯(1.72ml,12.79mmol)的thf(10ml)溶液,将反应混合物升温至大约21℃并搅拌16小时。减压蒸发溶剂,固体残余物用乙醚研制,过滤并用乙醚洗涤。然后将固体用饱和nh4cl溶液(30ml)稀释,水相用etoac(2
×
200ml)萃取。有机层经na2so4干燥并浓缩。粗产物通过硅胶柱色谱法使用10%etoac/己烷纯化,得到化合物b2,其为黄色固体(2.06g,7.46mmol,64.3%)。
[0250]1h nmr(400mhz,cdcl3):δ8.75(d,j=5.2hz,1h),8.32(d,j=5.2hz,1h),8.08(d,j
=8.0hz,1h),7.89(t,j=7.6hz 1h),7.56-7.51(m,2h),7.28-7.25(m,1h),4.55(s,2h)
[0251]
lc-ms(esi):m/z 277[m]
[0252]
6.2.4.2-溴-4-[3-(吡啶-2-基)-1h-吡唑-4-基]吡啶(b3)
[0253]
在氩气下用在dmf中的冰乙酸(0.45ml,7.39mmol)处理在纯(dry)dmf(3.4ml)中的b2(850mg,3.07mmol)溶液。逐滴加入dma(0.6ml,4.61mmol)并将混合物在大约21℃的氩气氛下搅拌2小时。逐滴加入一水合肼(1.15ml,23.09mmol)并将所得混合物在50℃加热3小时并在大约21℃加热16小时。将反应混合物倒入水(30ml)中并用ch2cl2(3
×
30ml)萃取。有机层经na2so4干燥并过滤。减压蒸发溶剂,得到粗化合物。粗产物通过硅胶柱色谱法使用30%etoac/己烷纯化,得到化合物b3,其为黄色固体(560mg,1.86mmol,60.6%)。
[0254]1h nmr(400mhz,cdcl3)δ8.74(brs,1h),8.34(d,j=5.0hz,1h),7.83(brs,1h),7.81(t,j=6.0hz,1h),7.56(s,1h),7.49(d,j=8.0hz,1h),7.39-7.84(m,1h),7.31-7.26(m,1h)
[0255]
lc-ms(esi):m/z 301[m]
[0256]
6.2.5.2-溴-4-(3-(吡啶-2-基)-1-三苯甲基-1h-吡唑-4-基)吡啶(b4)
[0257]
向b3(500mg,1.66mmol)在丙酮(10ml)中的搅拌溶液中加入k2co3(1.37g,9.99mmol)和三苯甲基氯(464mg,2.49mmol)。随后将反应混合物加热至回流并搅拌24小时。过滤反应混合物并浓缩滤液,然后在ch2cl2(20ml)和水(10ml)之间分配。有机相经na2so4干燥并浓缩。粗固体通过硅胶柱色谱法使用2%meoh/ch2cl2纯化,得到化合物b4,其为淡黄色固体(402mg,0.74mmol,44%)。
[0258]1h nmr(500mhz,cdcl3):δ8.53(d,j=4.5hz,1h),8.20(d,j=5.5hz,1h),7.75-7.05(m,2h),7.56(s,1h),7.51(s,1h),7.35-7.32(m,9h),7.25-7.22(m,8h)
[0259]
6.2.6.甲基(2-(4-(4-(3-(吡啶-2-基)-1-三苯甲基-1h-吡唑-4-基)吡啶-2-基)苯氧基)乙基)氨基甲酸叔丁酯(b5)
[0260]
在氩气氛下向b4(100mg,0.18mmol)在甲苯(2ml)中的搅拌溶液中加入在etoh(0.75ml)中的int-b(185mg,0.49mmol),然后加入2m na2co3溶液(0.45ml)。将反应混合物用氩气脱气20分钟,然后加入pd(pph3)4(16mg,0.01mmol)并回流3小时。在起始材料完全消耗后(通过tlc监测),将反应混合物倒入水中并用甲苯(3
×
15ml)萃取。有机层经na2so4干燥并减压浓缩,得到粗产物,将其通过硅胶柱色谱法使用30%etoac/己烷纯化,得到化合物b5,其为无色固体(70mg,0.09mmol,53%)。
[0261]1h nmr(400mhz,cdcl3):δ8.53(s,1h),8.49(d,j=4.8hz,1h),7.82(d,j=8.8hz,2h)7.74-7.76(m,3h),7.60(s,1h),7.40-7.34(s,8h),7.31-7.30(m,2h),7.24-7.19(m,4h),7.12-7.10(m,1h),6.93(d,j=8.8hz,2h),4.19-4.12(m,2h),3.66-3.58(m,2h),2.98(s,3h),1.46(s,9h)。
[0262]
6.2.7.n-甲基-2-(4-(4-(3-(吡啶-2-基)-1h-吡唑-4-基)吡啶-2-基)苯氧基)乙烷-1-胺盐酸盐(化合物b)
[0263]
在0℃,向在ch2cl2(6ml)中的搅拌的溶液b5(70mg,0.09mmol)中加入1,4-二恶烷(0.5ml)中的4n hcl。将反应混合物在氩气氛下搅拌1小时。起始材料完全消耗后(通过tlc监测),减压蒸发溶剂,得到粗化合物,用正戊烷(2x1ml)研磨,干燥,得到化合物b盐酸盐,其为无色固体(25mg,0.06mmol,69%)。
[0264]1h nmr(400mhz,dmso-d6):δ8.94(brs,2h),8.62-8.56(m,3h),8.30(brs,1h),8.03-7.96(m,3h),7.86(d,j=7.6hz,1h),7.69(brs,1h),7.49(dd,j=7.2,5.6hz,1h),7.29(d,j=7.6hz,1h),7.20(d,j=8.4hz,1h),4.36(t,j=4.8hz,2h),3.39-3.35(m,2h),2.67-2.63(m,3h)
[0265]
lc-ms(esi):m/z 372[m h]
[0266]
6.3.实施例3:合成n-甲基-2-(4-{4-[3-(6-甲基吡啶-2-基)-1h-吡唑-4-基]吡啶-2-基}苯氧基)乙烷-1-胺(化合物c)
[0267]
化合物c根据以下方案3中的通用方法制备:
[0268][0269]
6.3.1.2-(2-溴吡啶-4-基)-1-(6-甲基吡啶-2-基)乙烷-1-酮(c2)
[0270]
在-78℃在氩气下,向2-溴-4-甲基吡啶(b1)(1g,5.81mmol)在thf(15ml)中的搅拌溶液中逐滴加入nahmds(在thf中2m,6.39ml,12.8mmol)。将黄色溶液在-78℃下搅拌30分钟。然后加入6-甲基吡啶甲酸甲酯(1.19ml,8.72mmol)在thf(7ml)中的溶液,使反应混合物升温至达约21℃并搅拌16小时。减压蒸发溶剂,固体残余物用乙醚研制,过滤并用乙醚洗涤。然后将固体用饱和nh4cl溶液(20ml)稀释,水相用etoac(2
×
150ml)萃取。有机层经na2so4干燥并浓缩。粗产物通过硅胶柱色谱法使用10%etoac/己烷纯化,得到化合物c2,其为黄色固体(1.1g,3.79mmol,65.4%)。
[0271]1h nmr(500mhz,cdcl3):δ8.30(d,j=5.0hz,1h),7.86(d,j=8hz,1h),7.73(t,j=7.5hz,1h),7.51(s,1h),7.36(d,j=8hz,1h),7.24(d,j=5hz,1h),4.52(s,2h),2.64(s,3h)
[0272]
lc-ms(esi):m/z 291[m]
[0273]
6.3.2.2-溴-4-[3-(6-甲基吡啶-2-基)-1h-吡唑-4-基]吡啶(c3)
[0274]
在氩气下,用在dmf中的冰乙酸(0.14ml,2.48mmol)处理在纯dmf(1ml)中的c2(300mg,1.03mmol)溶液。逐滴加入dma(0.2ml,1.55mmol)并将混合物在大约21℃在氩气氛下搅拌1小时。逐滴加入一水合肼(0.37ml,7.75mmol)并将所得混合物在50℃加热3小时并在大约21℃加热16小时。将反应混合物倒入水(20ml)中并用ch2cl2(3
×
20ml)萃取。有机层
经na2so4干燥并过滤。减压蒸发溶剂,得到粗c3。粗c3通过硅胶柱色谱法使用2%meoh/dcm纯化,得到纯化的c3,其为黄色固体(172mg,0.54mmol,53%)。
[0275]1h nmr(500mhz,cdcl3):δ11.40(brs,1h),8.37(d,j=5.0hz,1h),7.74(s,1h),7.64(s,1h),7.58(t,j=8.0hz,1h),7.34(d,j=6.0hz,1h),7.26(d,j=8.0hz,1h),7.17(d,j=8.0hz,1h),2.60(s,3h)
[0276]
lc-ms(esi):m/z 315[m]
[0277]
6.3.3.2-溴-4-(3-(6-甲基吡啶-2-基)-1-三苯甲基-1h-吡唑-4-基)吡啶(c4)
[0278]
向在丙酮(2ml)中的c3(40mg,0.12mmol)的搅拌溶液中加入k2co3(53mg,0.38mmol)和三苯甲基氯(53mg,0.19mmol)。随后将反应混合物加热至回流并搅拌24小时。过滤反应混合物并浓缩滤液,然后在ch2cl2(5ml)和水(5ml)之间分配。有机相用na2so4干燥并浓缩。粗固体通过硅胶柱色谱法使用2%meoh/ch2cl2纯化,得到化合物c4,其为淡黄色固体(30mg,0.05mmol,41%)。
[0279]1h nmr(400mhz,cdcl3):δ8.22(d,j=4.8hz,1h),7.73(s,1h),7.59(s,3h),7.39-7.35(m,9h),7.31(s,1h),7.28-7.25(m,6h),7.24(d,j=12hz,1h),2.53(s,3h)
[0280]
lc-ms(esi):m/z 558[m]
[0281]
6.3.4.甲基(2-(4-(4-(3-(6-甲基吡啶-2-基)-1-三苯甲基-1h-吡唑-4-基)吡啶-2-基)苯氧基)乙基)氨基甲酸叔丁酯(c5)
[0282]
在氩气氛下向在甲苯(5ml)中的c4(150mg,0.26mmol)搅拌溶液中加入int-b(152mg,0.40mmol)在etoh(1ml)中的溶液,然后加入2m na2co3溶液(0.7ml)。将反应混合物用氩气脱气20分钟,然后加入pd(pph3)4(25mg,0.02mmol)并回流6小时。在起始材料完全消耗后(通过tlc监测),将反应混合物倒入水中并用甲苯(3
×
10ml)萃取。有机层经na2so4干燥并减压浓缩,得到粗c5,将其通过硅胶柱色谱法使用30%etoac/己烷纯化,得到纯化的c5,其为棕色固体(51mg,0.07mmol,26%)。
[0283]1h nmr(400mhz,cdcl3):δ8.48(d,j=5.2hz,1h),7.82(d,j=8.8hz,3h),7.74(s,1h),7.60(s,1h),7.56(d,j=15.2hz,j=7.6hz,2h),7.35-7.33(m,8h),7.28-7.27(m,6h),7.08(d,j=6.8hz,2h),6.93(d,j=8.8hz,2h),4.16-4.08(m,2h),3.63-3.58(m,2h),2.98(s,3h),2.41(s,3h),1.46(s,9h)
[0284]
6.3.5.n-甲基-2-(4-{4-[3-(6-甲基吡啶-2-基)-1h-吡唑-4-基]吡啶-2-基}苯氧基)乙烷-1-胺(化合物c)
[0285]
在0℃下向在ch2cl2(5ml)中的c5(51mg,0.07mmol)搅拌溶液中加入1,4-二恶烷(0.3ml)中的4n hcl。然后将反应混合物在氩气氛下搅拌1小时。在起始材料完全消耗后(通过tlc监测),减压蒸发溶剂以获得粗化合物c。然后将粗化合物c与正戊烷(2x1ml)一起研磨并干燥,得到化合物c,其为hcl盐,为棕色固体(20mg,0.05mmol,74%)。
[0286]1h nmr(400mhz,dmso-d6):δ8.93(brs,2h),8.61(d,j=5.6hz,1h),8.56(brs,1h),8.33(brs,1h),8.03(d,j=8.8hz,2h),7.88(t,j=7.6hz,1h),7.78-7.74(m,1h),7.65(d,j=7.2hz,1h),7.38(d,j=7.6hz,1h),7.20(d,j=8.4hz,2h),4.36(t,j=5.2hz,2h),3.36(t,j=5.2hz,2h),2.66-2.63(m,3h),2.50-2.46(m,3h)
[0287]
lc-ms(esi):m/z 386[m]
[0288]
6.4.实施例4:合成(z)-n-乙基-3-(((4-(n-(2-(甲基氨基)乙基)甲基磺酰胺基)
苯基)氨基)(苯基)亚甲基)-2-氧代吲哚啉-6-甲酰胺((z)-n-ethyl-3-(((4-(n-(2-(methylamino)ethyl)methylsulfonamido)phenyl)amino)(phenyl)methylene)-2-oxoindoline-6-carboxamide)(化合物d)
[0289]
化合物d根据以下方案4中的一般方法制备:
[0290][0291]
6.4.1.1-乙酰基-2-氧代吲哚啉-6-羧酸甲酯(d2)
[0292]
在惰性气氛下加热在乙酸酐(16ml)中的2-氧代吲哚啉-6-羧酸甲酯(d1)(2.0g,10.47mmol)的搅拌溶液至130℃达6h。在起始材料完全消耗(通过tlc监测)后,将反应混合物冷却至大约21℃。过滤沉淀物,用正己烷(2x50ml)洗涤并真空干燥,得到化合物d2,其为黄色固体(1.5g,61.5%)。
[0293]1h nmr(400mhz,dmso-d6):δ8.66(s,1h),7.82(d,j=8.0hz,1h),7.48(d,j=8.0hz,1h),3.91(s,2h),3.87(s,3h),2.57(s,3h)
[0294]
6.4.2.(z)-1-乙酰基-3-(羟基(苯基)亚甲基)-2-氧代吲哚啉-6-羧酸甲酯(d3)
[0295]
在0℃,惰性气氛下向化合物d2(1.5g,6.43mmol)在dmf(10ml)中的搅拌溶液中加入tbtu(2.69g,8.36mmol)、苯甲酸(903mg,7.40mmol)和三乙胺(2.2ml)。将反应混合物升温
至大约21℃并搅拌16小时。在起始材料完全消耗后(通过tlc监测),反应混合物用冰冷的水(30ml)淬灭并用etoac(2
×
40ml)萃取。合并的有机萃取物经na2so4干燥,过滤并真空浓缩,得到粗产物d3,将其通过硅胶柱色谱法使用80%etoac/己烷纯化,得到化合物d3(900mg,42%),其为黄色固体。
[0296]1h nmr(400mhz,cdcl3):δ14.01(brs,1h),8.93(s,1h),7.76-7.70(m,3h),7.67-7.63(m,1h),7.59-7.56(m,2h),7.12(d,j=8.0hz,1h),3.90(s,3h),2.83(s,3h)
[0297]
lc-ms(esi):m/z 338.3[m h]
[0298]
6.4.3.(z)-3-(羟基(苯基)亚甲基)-2-氧代吲哚啉-6-羧酸(d4)
[0299]
在大约21℃下向化合物d3(900mg,2.67mmol)在meoh(15ml)中的搅拌溶液中加入1n水性naoh溶液(15ml)。将混合物加热至100℃并搅拌6小时。在起始材料完全消耗(通过tlc监测)后,将反应混合物冷却至大约21℃,用1n水性hcl溶液(13ml)淬灭,并搅拌30分钟。过滤沉淀的固体,用20%etoac/己烷洗涤,得到化合物d4(580mg,77%),其为灰白色固体,无需进一步纯化即可用于下一步。
[0300]1h nmr(400mhz,dmso-d6):δ12.76(brs,1h),11.61(brs,1h),7.77-7.50(m,8h),7.13(brs,1h)
[0301]
6.4.4.(z)-n-乙基-3-(羟基(苯基)亚甲基)-2-氧代吲哚啉-6-碳酰胺化物((z)-n-ethyl-3-(hydroxy(phenyl)methylene)-2-oxoindoline-6-carboxamidelate)(片段a)
[0302]
在大约21℃,惰性气氛下向化合物d4(580mg,2.06mmol)在dmf(10ml)中的搅拌溶液中加入tbtu(729mg,2.27mmol)、hobt(306mg,2.27mmol)和n,n-二异丙基乙胺(1.9ml,10.32mmol)。30分钟后,在0℃下加入在thf(2.1ml,4.12mmol)中的2n乙胺并搅拌1小时。然后将反应混合物升温至大约21℃并再搅拌16小时。在起始材料完全消耗后(通过tlc监测),真空除去挥发物。将残余物用水(15ml)稀释,过滤并用20%etoac/己烷(2x10ml)洗涤以获得粗产物,将其通过硅胶柱色谱法使用10%meoh/ch2cl2纯化至得到片段a(410mg,64.5%),其为灰白色固体。
[0303]1h nmr(400mhz,dmso-d6):δ13.62(brs,1h),11.39(brs,1h),8.35-8.33(m,1h),7.76-7.52(m,5h),7.44-7.36(m,3h),3.29-3.22(m,2h),1.10(t,j=7.2hz,3h)
[0304]
lc-ms(esi):m/z 307.1(m-h
)
[0305]
6.4.5.n-(2-(二甲基氨基)乙基)-n-(4-硝基苯基)甲磺酰胺(d8)
[0306]
在0℃,惰性气氛下向化合物d7(800mg,3.70mmol)在丙酮(15ml)中的搅拌溶液中加入碳酸钾(1.32g,9.62mmol)、碘化钠(110mg,0.74mmol)和化合物b6(799mg,5.55mmol)。将反应混合物加热至50℃并搅拌20小时。在起始材料完全消耗后(通过tlc监测),真空除去挥发物。残余物用水(20ml)稀释并用etoac(2
×
40ml)萃取。合并的有机萃取物经na2so4干燥,过滤并真空浓缩,得到粗产物,将其通过硅胶柱色谱法使用5%meoh/ch2cl2纯化,得到化合物d8(460mg,43%),其为淡黄色固体。
[0307]1h nmr(500mhz,dmso-d6):δ8.27(d,j=9.5hz,2h),7.68(d,j=9.5hz,2h),3.85(t,j=6.5hz,2h),3.13(s,3h),2.31(t,j=6.5hz,2h),2.12(s,6h)
[0308]
lc-ms(esi):m/z 288.3(m-h
)
[0309]
6.4.6. n-(4-氨基苯基)-n-(2-(二甲基氨基)乙基)甲磺酰胺(片段b)
[0310]
向化合物d8(460mg,1.60mmol)在meoh(10ml)中的搅拌的溶液中加入10%pd/c
(40mg)并在大约21℃,氢气气氛(气球压力)下搅拌3小时。在起始材料完全消耗后(通过tlc监测),将反应混合物通过垫过滤并用meoh(10ml)洗涤。将滤液真空浓缩,得到粗产物,将其通过硅胶柱色谱法使用10%meoh//ch2cl2纯化,得到片段b(300mg 73%),其为淡黄色固体。
[0311]1h nmr(400mhz,dmso-d6):δ6.99(d,j=8.8hz,2h),6.54(d,j=8.8hz,2h),5.25(s,2h),3.55(t,j=7.2hz,2h),2.91(s,3h),2.24(t,j=7.2hz,2h),2.12(s,6h)
[0312]
lc-ms(esi):m/z 258.2(m-h
)
[0313]
6.4.7.(z)-3-(((4-(n-(2-(二甲基氨基)乙基)甲基磺酰胺基)苯基)氨基)(苯基)亚甲基)-n-乙基-2-氧代吲哚啉-6-甲酰胺(d5)
[0314]
将在thf(5ml)中片段a(200mg,0.64mmol)、片段b(500mg,1.94mmol)和tms-咪唑(455mg,3.24mmol)的溶液在微波下加热至170℃,1小时。在起始材料消耗后(通过tlc和lc-ms监测)后,真空除去挥发物。将残余物用水(10ml)稀释并用etoac(3
×
25ml)萃取,得到粗产物,将其通过制备型hplc纯化,得到化合物d5(150mg,42%),其为淡黄色固体。
[0315]1h nmr(400mhz,dmso-d6):δ12.14(s,1h),10.91(s,1h),8.17(t,j=5.6hz,1h),7.64-7.57(m,3h),7.53-7.51(m,2h),7.34(s,1h),7.17(d,j=8.8hz,2h),7.06(d,j=8.4hz,1h),6.84(d,j=8.8hz,2h),5.73(d,j=8.4hz,1h),3.58(t,j=6.8hz,2h),3.23-3.20(m,2h),2.93(s,3h),2.13(t,j=6.8hz,2h),1.90(s,6h),1.06(t,j=7.2hz,3h)
[0316]
lc-ms(esi):m/z 548.6(m-h
)
[0317]
6.4.8.(z)-n-乙基-3-(((4-(n-(2-(甲基氨基)乙基)甲基磺酰胺基)苯基)氨基)(苯基)亚甲基)-2-氧代吲哚啉-6-甲酰胺(化合物d)
[0318]
在大约21℃,在惰性气氛下,向化合物d5(70mg,0.12mmol)在纯甲苯(3ml)中的搅拌溶液中加入2,2,2-三氯乙氧基碳酰氯(0.04ml,0.19mmol)。将反应混合物加热至回流温度(120℃)并保持16小时。起始材料消耗(通过tlc监测)后,将反应混合物冷却至大约21℃,用etoac(30ml)稀释并用1n水性hcl溶液(15ml)洗涤。有机层经na2so4干燥,过滤并真空浓缩,得到单脱甲基化的、用di-troc-保护的化合物(40mg)。
[0319]
在大约21℃,惰性气氛下将来自上述反应的粗产物在乙酸(3ml)中溶解并加入锌粉末(9mg,0.13mmol)。将反应混合物加热至50℃并搅拌8小时。在起始材料完全消耗后(通过tlc监测),将反应混合物冷却至约21℃并真空除去挥发物。残余物用水(20ml)稀释并用etoac(2
×
25ml)萃取。合并的有机萃取物用饱和nahco3溶液(20ml)洗涤,经na2so4干燥,过滤并减压浓缩以获得粗化合物d,将其通过硅胶柱色谱法使用5-6%meoh/ch2cl2纯化,得到12mg化合物d,hplc纯度为83%。
[0320]
以60mg规模重复反应,将获得的粗产物与上述批次合并,通过制备型hplc纯化,得到化合物d(8.0mg,6.3%),其为淡黄色固体。
[0321]1h nmr(400mhz,cd3od):δ7.65-7.59(m,3h),7.52.7.50(m,2h),7.40(s,1h),7.31(d,j=8.8hz,2h),7.07(d,j=8.4hz,1h),6.90(d,j=8.8hz,2h),5.95(d,j=8.4hz,1h),3.95(t,j=5.6hz,2h),3.39-3.32(m,2h),3.05(t,j=5.6hz,2h),2.93(s,3h),2.71(s,3h),1.19(t,j=7.2hz,3h)
[0322]
lc-ms(esi):m/z 534.6(m-h
)
[0323]
uplc纯度:99.18%
[0324]
6.5.实施例5:(z)-n-乙基-3-(((4-(n-(2-(甲基氨基)乙基)甲基磺酰胺基)苯基)氨基)(苯基)亚甲基)-2-氧代吲哚啉-6-甲酰胺(化合物d)的替代合成
[0325]
化合物d也根据以下方案5中的一般方法制备:
[0326][0327]
6.5.1.n-(2-溴乙基)-n-(4-硝基苯基)甲磺酰胺(d9)
[0328]
在0℃,惰性气氛下向化合物d7(1.0g,4.65mmol)在dmf(10ml)中的搅拌溶液中加入氢化钠(60%在矿物油中;320mg,7.99mmol),并在大约21℃下搅拌30分钟。在大约21℃下向该混合物中加入1,2-二溴乙烷(2.18g,11.60mmol)。将混合物加热至90℃并搅拌24小时。通过tlc监测反应。将反应混合物冷却至大约21℃,用冰冷的水(30ml)淬灭并用etoac(2x40ml)萃取。将合并的有机萃取物用na2so4干燥,过滤并真空浓缩,得到粗产物,将其通过硅胶柱色谱法使用5%meoh/ch2cl2纯化,得到1.2g d9,其为含有40%未反应的起始材料的混合物。所得混合物不经进一步纯化直接用于下一步反应。
[0329]1h nmr(500mhz,cdcl3):δ8.29(d,j=8.5hz,2h),7.56(d,j=8.5hz,2h),4.12(t,j=7.0hz,2h),3.44(t,j=7.0hz,2h),3.01(s,3h)
[0330]
6.5.2.n-(2-(甲基氨基)乙基)-n-(4-硝基苯基)甲磺酰胺(d10)
[0331]
在大约21℃下惰性气氛下的密封管中向化合物d9(1.2g,不纯)在thf(10ml)中的混合溶液中加入三乙胺(1.6ml)和甲胺(在thf中2m;9.3ml,18.63mmol)。将反应混合物加热
至80℃并保持16小时。在起始材料完全消耗后(通过tlc监测),将反应混合物冷却至大约21℃并减压浓缩以获得粗d10。粗d10通过硅胶柱色谱法使用15%meoh/ch2cl2纯化,得到化合物d10,其为黄色固体(500mg,两步中的总产率为39%)。
[0332]1h nmr(500mhz,dmso-d6)δ8.94(brs,1h),8.31(d,j=9.0hz,2h),7.80(d,j=8.5hz,2h),4.06(t,j=6.0hz,2h),3.15(s,3h),3.00(t,j=6.0hz,2h),2.55(s,3h)
[0333]
6.5.3.甲基(2-(n-(4-硝基苯基)甲基磺酰胺基)乙基)氨基甲酸叔丁酯(d11)
[0334]
在大约21℃,惰性气氛下,向d10(500mg,1.83mmol)在ch2cl2(10ml)中的搅拌溶液中加入三乙胺(0.4ml,2.61mmol)和boc-酸酐(659mg,3.02mmol)并保持5小时。在起始材料完全消耗后(通过tlc监测),在真空中除去挥发物以获得粗产物,将其通过硅胶柱色谱法使用5%meoh/ch2cl2纯化,得到d11,其为无色粘稠糖浆(320mg,47%)。
[0335]1h nmr(400mhz,dmso-d6):δ8.27(d,j=8.4hz,2h),7.68(d,j=8.4hz,2h),3.91(t,j=6.4hz,2h),3.28-3.25(m,2h),3.07(s,3h),2.72-2.70(m,3h),1.33-1.27(m,9h)
[0336]
lc-ms(esi):m/z 274.2(m
-b℃)
[0337]
6.5.4.(2-(n-(4-氨基苯基)甲基磺酰胺基)乙基)(甲基)氨基甲酸叔丁酯(片段b的boc-变体)
[0338]
向化合物d11(250mg,0.67mmol)在etoh(10ml)中的溶液中加入雷尼(raney)-ni(40mg)并在氢气氛(气球压力)下在大约21℃下搅拌1小时。在起始材料完全消耗后(通过tlc监测),将反应混合物通过垫过滤并用etoh(10ml)洗涤。将合并的滤液真空浓缩以获得粗产物,将其通过硅胶柱色谱法使用10%meoh/ch2cl2纯化,得到片段b的boc-变体,其为淡黄色固体(180mg,77%)。
[0339]1h nmr(400mhz,dmso-d6):δ7.01(d,j=8.4hz,2h),6.53(d,j=8.4hz,2h),5.24(s,2h),3.60(t,j=6.4hz,2h),3.18(t,j=6.4hz,2h),2.88(s,3h),2.75-2.71(m,3h),1.36-1.33(m,9h)
[0340]
lc-ms(esi):m/z 244.2(m
-b℃)
[0341]
6.5.5.(z)-(2-(n-(4-(((6-(乙基氨基甲酰基)-2-氧代吲哚啉-3-亚基)(苯基)甲基)氨基)苯基)甲基磺酰胺基)乙基)(甲基)氨基甲酸叔丁酯(tert-butyl
[0342]
(z)-(2-(n-(4-(((6-(ethylcarbamoyl)-2-oxoindolin-3-ylidene)(phenyl)methyl)amino)phenyl)methylsulfonamido)ethyl)(methyl)carbamate)(d10)
[0343]
片段a(70mg,0.22mmol)、片段b的boc-变体(155mg,0.45mmol)和tms-咪唑(159mg,1.13mmol)在thf(3ml)中的溶液在微波下加热至170℃,160分钟。在起始材料消耗后(通过tlc和lc-ms监测),真空除去挥发物以获得残余物,将其通过制备型hplc纯化,得到化合物d10(50mg,36%),其为淡黄色固体。
[0344]1h nmr(400mhz,cdcl3):δ12.13(brs,1h),8.01(brs,1h),7.61-7.51(m,3h),7.44-7.41(m,3h),7.13-7.11(m,2h),6.98(d,j=8.4hz,1h),6.75(d,j=8.4hz,2h),5.96-5.91(m,2h),3.74-3.71(m,2h),3.49-3.41(m,2h),3.30-3.27(m,2h),2.80(s,6h),1.40-1.36(m,9h),1.19(t,j=7.2hz,3h)
[0345]
lc-ms(esi):m/z 634.6[m h]
[0346]
6.5.6.(z)-n-乙基-3-(((4-(n-(2-(甲基氨基)乙基)甲基磺酰胺基)苯基)氨基)(苯基)亚甲基)-2-氧代吲哚啉-6-甲酰胺氢氯化物(化合物d作为hcl盐)
[0347]
在0℃,惰性气氛下,向化合物d10(20mg,0.03mmol)在乙醚(3ml)中的搅拌溶液中加入在1,4-二恶烷(0.3ml)中的4n hcl。将反应混合物在约21℃搅拌1小时。在起始材料完全消耗后(通过tlc监测),在真空中除去挥发物以获得粗产物,将其与正戊烷(2x4ml)一起研磨,得到hcl盐形式的化合物d(12mg,71%),其为淡黄色固体。
[0348]1h nmr(400mhz,cd3od):δ7.65-7.59(m,3h),7.52.7.50(m,2h),7.40(s,1h),7.31(d,j=8.8hz,2h),7.07(d,j=8.4hz,1h),6.90(d,j=8.8hz,2h),5.95(d,j=8.4hz,1h),3.95(t,j=5.6hz,2h),3.39-3.32(m,2h),3.05(t,j=5.6hz,2h),2.93(s,3h),2.71(s,3h),1.19(t,j=7.2hz,3h)。
[0349]
lc-ms(esi):m/z 534.7[m h]
[0350]
uplc纯度:96.26%
[0351]
6.6.实施例6:测试化合物a-d活性的体外测定
[0352]
测试化合物a-d以确定它们是否可以在体外在hek293t细胞中抑制tgf-β诱导的萤光素酶活性。
[0353]
将30,000个hek293t细胞接种在96孔白色平底板中过夜。第二天,使用lipofectamine 24小时将每孔100ng的smad萤光素酶报告质粒转染到细胞中。第二天用化合物a-d和100pm的tgf-β处理细胞24小时。使用dual-glo萤光素酶测定试剂盒(promega)测量萤光素酶活性。化合物a、b和d进行了两次测定,化合物c进行了3次。表4中显示结果。
[0354][0355]
实验1的活性数据显示在图1中。
[0356]
化合物a-c展示出最大的抑制活性。
[0357]
6.7.实施例7:合成4-((s)-2-((s)-2-(6-(2,5-二氧代-2h-吡咯-1(5h)-基)六酰胺基)-3-甲基丁酰胺基)-5-脲基戊酰胺基)苄基甲基(2-(4-(4-(3-(6-甲基吡啶-2-基)-1h-吡唑-4-基)吡啶-2-基)苯氧基)乙基)氨基甲酸酯
[0358]
根据以下方案6中的通用方法,将化合物c与缬氨酸-瓜氨酸接头连接:
[0359][0360]
在0℃下将l1(122mg,0.165mmol,1.1当量)和tea(52μl,0.375mmol,2.5当量)添加到化合物c(58mg,0.150mmol,1.0当量)在dmf(2ml)中的溶液中,将反应混合物在约21℃下搅拌2小时,得到粗adc-1。通过制备型hplc纯化粗adc-1,得到纯化的adc-1,其为白色固体(34mg,24%产率)。
[0361]
6.8.实施例8:生成抗体药物缀合物1(adc1)
[0362]
将抗人fap抗体透析到缀合缓冲液(25mm硼酸钠/25mm nacl,和0.3mm edta,最终ph 7.4)中过夜。使用三(2-羧乙基)膦(tcep)以10-30的还原比还原抗体2小时。adc-1溶解在dmso中至终浓度为10mm,然后在15%dmso存在下以5-30的缀合比与抗体缀合。所有反应均在约21℃下进行。对于一些药物抗体比(dar),在缀合步骤中使用50%的丙二醇作为有机溶剂。最终的adc在pbs中透析过夜,使用0.22μm过滤器过滤并经由hplc-hic分析以确定dar和hplc-sec以确定聚集水平。对于hplc-hic,样品在tskgel丁基-npr柱上以0.5ml/min的流速运行。a相是25mm的磷酸钠和1.5m的硫酸铵,ph 6.95,而b相是ph 6.95的75%的25mm磷酸钠和25%的异丙醇。对于hplc-sec分析,在流速为0.25ml/min,以280nm使用g3000sw柱(tosoh bioscience),达25分钟。
[0363]
6.9.实施例9:合成与二硫化物接头(adc-2)连接的化合物c
[0364]
根据以下方案7a-b中的通用方法,将化合物c连接到二硫化物接头:
[0365]
[0366][0367]
6.9.1.合成中间物a
[0368]
2-氯三苯甲基氯树脂(l2)(4g,4mmol)用dcm(2x40ml)洗涤,在50ml的dcm中溶胀10
吡唑-3-基)吡啶-2-1)苯氧基)乙基)氨基甲酰氧基)乙基)二硫烷基)甲基)-4,7,10,13,16-五氧代-3,6,9,12,15-五氮杂二十烷-1,20-二酸(l15)
[0377]
向l13(32mg,0.025mmol)在dmf(3ml)中的溶液中加入2,5-二氧代吡咯烷-1-基2-(叔丁氧基羰基氨基氧基)乙酸酯(l14)(28mg,0.097mmol),然后加入tea(0.5ml)。将反应混合物在n2气氛下在大约21℃搅拌16小时以提供l15。通过制备型hplc纯化粗l15,得到12mg纯化的l15,其为白色固体(产率33%)
[0378]
6.9.5.(2r,5s,8s,11s,14s,19s)-19-(2-(氨基氧基)乙酰氨基)-5,8,14-三(羧甲基)-11-(3-胍基丙基)-2-(((2-(甲基(2-(4-(4(4-(6-甲基吡啶-2-基)-1h-吡唑-3-基)吡啶-2-基)苯氧基)乙基)氨基甲酰氧基)乙基)二硫烷基)甲基)-4,7,10,13,16-五氧代-3,6,9,12,15-五氮杂二十烷-1,20-二酸(adc-2)
[0379]
l15(12mg,0.0085mmol)在dcm(5ml)中的混合物中加入tfa(1ml)。将混合物在大约21℃下搅拌30分钟以提供adc-2。将粗adc-2浓缩并用制备型hplc纯化,得到3.5mg纯化的adc-2,其为白色固体(产率31%)。
[0380]
6.10.实施例10:抗体药物偶联物2(adc2)的生成
[0381]
根据以下方案b的一般方法,经由抗体赖氨酸残基将adc-2与抗人fap抗体连接:
[0382][0383]
将抗体透析到ph 7.4的pbs中。将s-4fb以不同的摩尔比添加到ph7.4的pbs中的抗体中,并在大约21℃下温育3小时。将s-4fb修饰的抗体溶液与2-肼基吡啶溶液(0.5mm,在100mm mes缓冲液,ph5.0),并在37℃下以范围为5-50的各种缀合比温育30分钟。s4fb/ab摩尔取代比由a354处的uv-vis确定。使用zeba
tm
旋转脱盐柱纯化修饰的抗体,将缓冲液更换为50mm的磷酸盐缓冲液(ph 6.5,150mm nacl),然后在37℃下以不同的摩尔比与接头-ss-药物adc-2(10mm,在dmso中)混合24小时以提供adc2。第二天,adc2样品针对pbs透析过夜。过滤样品,然后经由hplc-sec、sds-page和lc-ms进行测试。
[0384]
如果hplc-sec检测到超过5%的adc2聚集,则聚集的组分由akta与sec柱(ge healthcare life sciences,superdex 200increase 10/300gl)分离,并通过hplc-sec再次分析。
[0385]
6.11.实施例11:化合物n的合成和表征
[0386]
化合物n根据以下方案9中的通用方法合成:
[0387]
[0388]
在多个体外测定中,将化合物n与化合物c进行了比较。它们在重组激酶测定中的ic50活性和它们的ki值的总结显示在表5中。表5还显示了化合物c在抑制人hek细胞中tgf-β信号传导中的活性。在激酶测定中发现化合物c比化合物n有效10倍。
[0389][0390]
6.12.实施例12:抗fap抗体结合hek细胞
[0391]
通过facs评估抗fap抗体(商业获得小鼠igg1,克隆427819)结合表达人fap的hek293细胞的能力。抗fap抗体与转染人fap cdna并在细胞表面表达的hek293细胞结合(图2c),但不与未转染人cdna的亲本hek细胞结合(图2b)。
[0392]
6.13.实施例13:靶向性药物缀合物syn-301和syn-302的生产
[0393]
合成了两种靶向性药物缀合物。在第一种中,使用mc-val-cit-pabc可切割接头将化合物c缀合至抗fap抗体(商业可获得小鼠igg1,克隆427819)(图3a)。这种靶向性药物缀合物在本文中称为syn-301。在第二种中,使用不可切割(mc)接头将化合物与抗fap抗体缀合(图3b)。这种靶向性药物缀合物在本文中称为syn-302。使用plrp-s柱(孔径,5μm粒径,1x50mm)通过反相液相色谱确定药物抗体比,使用g3000swxl柱通过尺寸排阻色谱确定聚集百分比。syn-301制剂测得的药物抗体比(dar)为5.5和4%的聚集,而syn-302制剂测得dar为5和3.9%的聚集。
[0394]
6.14.在表达人fap蛋白的hek细胞中评估syn-301和syn-302对tgf-β信号传导抑制
[0395]
为了评估syn-301和syn-302抑制tgf-β信号传导的能力,使用表达人fap的hek293ft细胞进行了测定。
[0396]
hek293ft细胞用编码人fap的构建体以及tgf-β响应性萤光素酶表达构建体(该构建体包含驱动萤光素酶报告基因luc2p表达的smad结合元件的三个拷贝)(pgl4.48[luc2p/sbe/hygro];promega)和表达对照构建体(编码花虫型(renilla)萤光素酶的pgl4.74;promega)(1μg:1μg:0.125μg/ml的细胞培养物)用mirus-lt1转染试剂瞬时转染。将细胞以350,000个细胞/ml接种在96孔板中(每孔100μl)。24小时后,用syn-301、syn-302、未缀合的抗fap抗体或未缀合的化合物c预处理细胞4小时。然后,加入1nm的tgf-β并将细胞温育3小时。然后测量萤光素酶表达(dual-glo萤光素酶检测系统,promega)。
[0397]
结果示于图4a-4b,有可切割接头的syn-301能够抑制表达fap的工程化hek细胞中的tgf-β信号传导(图4a),而未观察到syn-301对不表达fap的亲本hek细胞中的tgf-β信号传导有显著影响(图4b)。在该测定中观察到具有不可切割接头的syn-302不如syn-301有效
(图4a)。
[0398]
6.15.实施例15:syn-301和syn-302诱导的fap内化
[0399]
为了评估syn-301和syn-302在内源性表达fap的靶细胞上内化fap的能力,使用wi-38人肺成纤维细胞进行内化测定。
[0400]
wi-38细胞与抗fap抗体、syn-301、syn-302或同种型对照adc(与可切割的valcit-alk5抑制剂化合物c缀合的非特异性抗体对照)在4℃下温育30分钟以检测细胞表面fap表达。然后用冷pbs洗涤细胞两次以去除上清液中残留的抗体/抗体缀合物,然后在37℃温育3小时以诱导受体内化。温育3小时后,洗涤细胞并与pe缀合的大鼠抗小鼠二抗温育以检测剩余的细胞表面fap表达。将相对的fap表达与在4℃下与抗fap抗体或缀合物一起温育的wi-38细胞(作为总fap表达的测量)进行比较。
[0401]
结果示于图5a-5e和图6。50-60%的wi-38细胞表达fap(图5a-5e),并且抗fap抗体syn-301和syn-302能够与wi-38细胞中的fap结合并相当地内化(分别为63%、63%和52%)(图6)。
[0402]
6.16.实施例16:在wi-38细胞中的syn-301和syn-302功能表征
[0403]
iv型胶原蛋白(col4a1)、纤连蛋白(fn1)和富含亮氨酸重复序列15(leucine rich repeat containing 15,lrrc15)的表达增加是纤维化增加的标志物。wi-38人肺成纤维细胞用于评估syn-301和syn-302降低col4a1、fn1和lrrc15表达的能力。
[0404]
以50,000个细胞/ml,在24个孔中(每孔500μl)铺板wi-38人肺成纤维细胞并温育过夜。将细胞血清饥饿18小时以降低血清对tgfb-β调节基因的影响,然后用syn-301、syn-302、同种型对照adc、抗fap抗体或化合物c以1μg/ml进行预处理。添加tgf-β并将细胞温育19小时。然后将细胞刮入rlt缓冲液(qiagen)并使用qiagen rnaeasy试剂盒提取rna。逆转录rna成cdna,然后用taqman引物对col4a1、fn1和lrrc15进行qpcr。gapdh用作标准化剂。
[0405]
结果示于图7a-7b。syn-301部分阻断tgf-β诱导的基因响应,降低col4a1表达约25-30%(图7a),降低fn1表达大约20-25%(图7a)和降低lrrc15表达大约15-20%(图7b)。syn-302在阻断tgf-β信号传导方面具有更温和的反应,而未缀合的抗fap抗体和同种型对照adc不抑制tgf-β信号传导。
[0406]
7.具体实施方案
[0407]
本公开通过以下具体实施方案来举例说明。
[0408]
1.靶向性药物缀合物,其包含与靶向部分可操作地连接的alk5抑制剂,该靶向部分与在肌成纤维细胞、活化的成纤维细胞、向肌成纤维细胞转变的成纤维细胞或其组合的表面上表达的细胞表面分子结合。
[0409]
2.根据实施方案1的靶向性药物缀合物,其中所述靶向部分与肌成纤维细胞细胞表面分子结合。
[0410]
3.根据实施方案1或实施方案2的靶向性药物缀合物,其中所述靶向部分与活化的成纤维细胞细胞表面分子结合。
[0411]
4.根据实施方案1至3中任一项的靶向性药物缀合物,其中所述靶向部分与向肌成纤维细胞转变的成纤维细胞细胞表面分子结合。
[0412]
5.根据实施方案1至4中任一项的靶向性药物缀合物,其中所述alk5抑制剂具有至少20nm的ic
50
。
[0413]
6.根据实施方案1至5中任一项的靶向性药物缀合物,其中所述alk5抑制剂为咪唑类化合物、吡唑类化合物或噻唑类化合物。
[0414]
7.根据实施方案6的靶向性药物缀合物,其中所述alk5抑制剂是咪唑类化合物。
[0415]
8.根据实施方案6的靶向性药物缀合物,其中所述alk5抑制剂是吡唑类化合物。
[0416]
9.根据实施方案6的靶向性药物缀合物,其中所述alk5抑制剂是噻唑类化合物。
[0417]
10.根据实施方案6的靶向性药物缀合物,其中所述alk5抑制剂为咪唑类化合物,其为咪唑-苯并二氧杂环戊烯(imidazole-benzodioxol)化合物或咪唑-喹喔啉(imidazole-quinoxaline)化合物。
[0418]
11.根据实施方案10的靶向性药物缀合物,其中所述alk5抑制剂是咪唑-苯并二氧杂环戊烯化合物。
[0419]
12.根据实施方案10的靶向性药物缀合物,其中所述alk5抑制剂是咪唑-喹喔啉化合物。
[0420]
13.根据实施方案6的靶向性药物缀合物,其中所述alk5抑制剂为吡唑类化合物,其为吡唑-吡咯并化合物。
[0421]
14.根据实施方案6的靶向性药物缀合物,其中所述alk5抑制剂为咪唑-苯并二氧杂环戊烯化合物、咪唑-喹喔啉化合物、吡唑-吡咯并化合物或噻唑类化合物。
[0422]
15.根据实施方案1至4中任一项的靶向性药物缀合物,其中所述alk5抑制剂是化合物c。
[0423]
16.根据实施方案1至4中任一项的靶向性药物缀合物,其中所述alk5抑制剂是化合物n。
[0424]
17.根据实施方案1至16中任一项的靶向性药物缀合物,其中所述alk5抑制剂经由接头与所述靶向部分连接。
[0425]
18.根据实施方案17的靶向性药物缀合物,其中所述接头是含有peg的接头。
[0426]
19.根据实施方案17或实施方案18的靶向性药物缀合物,其中所述接头是多价接头。
[0427]
20.根据实施方案17至19中任一项的靶向性药物缀合物,其中所述接头是不可切割接头。
[0428]
21.根据实施方案20的靶向性药物缀合物,其中所述不可切割接头是n-马来酰亚胺甲基环己烷1-羧酸酯(n-maleimidomethylcyclohexane1-carboxylate)、马来酰亚胺己酰基(maleimidocaproyl)或巯基乙酰氨基己酰基(mercaptoacetamidocaproyl)接头。
[0429]
22.根据实施方案21的靶向性药物缀合物,其中所述不可切割接头是n-马来酰亚胺甲基环己烷1-羧酸酯接头。
[0430]
23.根据实施方案21的靶向性药物缀合物,其中所述不可切割接头是马来酰亚胺己酰基接头。
[0431]
24.根据实施方案21的靶向性药物缀合物,其中所述不可切割接头是巯基乙酰氨基己酰基接头。
[0432]
25.根据实施方案17至19中任一项的靶向性药物缀合物,其中所述接头是可切割接头。
[0433]
26.根据实施方案25的靶向性药物缀合物,其中所述可切割接头是肽接头。
[0434]
27.根据实施方案25的靶向性药物缀合物,其中所述可切割接头是二肽接头、二硫化物接头(disulfide linker)或腙接头。
[0435]
28.根据实施方案27的靶向性药物缀合物,其中所述可切割接头是二肽接头。
[0436]
29.根据实施方案26的靶向性药物缀合物,其中所述可切割接头是三肽接头。
[0437]
30.根据实施方案26的靶向性药物缀合物,其中所述可切割接头是四肽接头。
[0438]
31.根据实施方案30的靶向性药物缀合物,其中所述肽接头是甘氨酸-甘氨酸-苯丙氨酸-甘氨酸(gly-gly-phe-gly)接头。
[0439]
32.根据实施方案27的靶向性药物缀合物,其中所述可切割接头是二硫化物接头。
[0440]
33.根据实施方案27的靶向性药物缀合物,其中所述可切割接头是腙接头。
[0441]
34.根据实施方案27的靶向性药物缀合物,其中所述接头是蛋白酶敏感性缬氨酸-瓜氨酸二肽接头。
[0442]
35.根据实施方案27的靶向性药物缀合物,其中所述接头是蛋白酶敏感性苯丙氨酸-赖氨酸二肽接头。
[0443]
36.根据实施方案27的靶向性药物缀合物,其中所述接头是谷胱甘肽敏感性二硫化物接头。
[0444]
37.根据实施方案27的靶向性药物缀合物,其中所述接头是酸敏感性二硫化物接头。
[0445]
38.根据实施方案1至37中任一项的靶向性药物缀合物,其中所述alk5抑制剂经由位点特异性缀合与所述靶向部分缀合。
[0446]
39.根据实施方案38的靶向性药物缀合物,其中所述alk5抑制剂经由所述靶向部分上的一个或多个半胱氨酸、赖氨酸或谷氨酰胺残基缀合。
[0447]
40.根据实施方案39的靶向性药物缀合物,其中所述alk5抑制剂经由所述靶向部分上的一个或多个半胱氨酸残基缀合。
[0448]
41.根据实施方案39的靶向性药物缀合物,其中所述alk5抑制剂经由所述靶向部分上的一个或多个赖氨酸残基缀合。
[0449]
42.根据实施方案39的靶向性药物缀合物,其中所述alk5抑制剂经由所述靶向部分上的一个或多个谷氨酰胺残基缀合。
[0450]
43.根据实施方案38的靶向性药物缀合物,其中所述alk5抑制剂经由所述靶向部分上的一个或多个非天然氨基酸残基缀合。
[0451]
44.根据实施方案43的靶向性药物缀合物,其中所述一个或多个非天然氨基酸残基包含对乙酰苯丙氨酸(pacf)。
[0452]
45.根据实施方案43的靶向性药物缀合物,其中所述一个或多个非天然氨基酸残基包含对叠氮基甲基-l-苯丙氨酸(pamf)。
[0453]
46.根据实施方案43的靶向性药物缀合物,其中所述一个或多个非天然氨基酸残基包含硒半胱氨酸(sec)。
[0454]
47.根据实施方案38的靶向性药物缀合物,其中所述alk5抑制剂经由所述靶向部分上的一种或多种聚糖缀合。
[0455]
48.根据实施方案47的靶向性药物缀合物,其中所述一种或多种聚糖包含岩藻糖。
[0456]
49.根据实施方案47的靶向性药物缀合物,其中所述一种或多种聚糖包含6-硫代
岩藻糖。
[0457]
50.根据实施方案47的靶向性药物缀合物,其中所述一种或多种聚糖包含半乳糖。
[0458]
51.根据实施方案47的靶向性药物缀合物,其中所述一种或多种聚糖包含n-乙酰半乳糖胺(galnac)。
[0459]
52.根据实施方案47的靶向性药物缀合物,其中所述一种或多种聚糖包含n-乙酰葡糖胺(glcnac)。
[0460]
53.根据实施方案47的靶向性药物缀合物,其中所述一种或多种聚糖包含唾液酸(sa)。
[0461]
54.根据实施方案38至53中任一项的靶向性药物缀合物,其中所述alk5抑制剂经由接头缀合。
[0462]
55.根据实施方案1至54中任一项的靶向性药物缀合物,其中每个靶向部分分子的alk5抑制剂分子的平均数在1至30之间的范围内。
[0463]
56.根据实施方案1至54中任一项的靶向性药物缀合物,其中每个靶向部分分子的alk5抑制剂分子的平均数在1至20之间的范围内。
[0464]
57.根据实施方案1至54中任一项的靶向性药物缀合物,其中每个靶向部分分子的alk5抑制剂分子的平均数在1至15之间的范围内。
[0465]
58.根据实施方案1至54中任一项的靶向性药物缀合物,其中每个靶向部分分子的alk5抑制剂分子的平均数在2至12之间的范围内。
[0466]
59.根据实施方案1至54中任一项的靶向性药物缀合物,其中每个靶向部分分子的alk5抑制剂分子的平均数在4至15之间的范围内。
[0467]
60.根据实施方案1至54中任一项的靶向性药物缀合物,其中每个靶向部分分子的alk5抑制剂分子的平均数在6至12之间的范围内。
[0468]
61.根据实施方案1至54中任一项的靶向性药物缀合物,其中每个靶向部分分子的alk5抑制剂分子的平均数在2至8之间的范围内。
[0469]
62.根据实施方案1至61的靶向性药物缀合物,其中所述靶向部分是内化性的。
[0470]
63.根据实施方案1至62中任一项的靶向性药物缀合物,其中所述靶向部分包含抗体或抗体片段。
[0471]
64.根据实施方案63的靶向性药物缀合物,其中所述靶向部分包含抗体。
[0472]
65.根据实施方案64的靶向性药物缀合物,其中所述抗体是单克隆抗体。
[0473]
66.根据实施方案65的靶向性药物缀合物,其中所述抗体是人的或人源化的。
[0474]
67.根据实施方案66的靶向性药物缀合物,其中所述抗体是人的。
[0475]
68.根据实施方案66的靶向性药物缀合物,其中所述抗体是人源化的。
[0476]
69.根据实施方案63的靶向性药物缀合物,其中所述靶向部分包含抗体片段。
[0477]
70.根据实施方案69的靶向性药物缀合物,其中所述抗体片段是单克隆抗体的片段。
[0478]
71.根据实施方案70的靶向性药物缀合物,其中所述抗体片段是人或人源化抗体的片段。
[0479]
72.根据实施方案71的靶向性药物缀合物,其中所述抗体片段是人抗体的片段。
[0480]
73.根据实施方案71的靶向性药物缀合物,其中所述抗体片段是人源化抗体的片
段。
[0481]
74.根据实施方案69至73中任一项的靶向性药物缀合物,其中所述抗体片段是fab、fab'、f(ab')2、fv、scfv、dsfv或单域抗体。
[0482]
75.根据实施方案74的靶向性药物缀合物,其中所述抗体片段是fab。
[0483]
76.根据实施方案74的靶向性药物缀合物,其中所述抗体片段是fab'。
[0484]
77.根据实施方案74的靶向性药物缀合物,其中所述抗体片段是f(ab')2。
[0485]
78.根据实施方案74的靶向性药物缀合物,其中所述抗体片段是fv。
[0486]
79.根据实施方案74的靶向性药物缀合物,其中所述抗体片段是scfv。
[0487]
80.根据实施方案79的靶向性药物缀合物,其中所述scfv包含scfv的vh和vl域之间的多肽接头。
[0488]
81.根据实施方案74的靶向性药物缀合物,其中所述抗体片段是dsfv。
[0489]
82.根据实施方案74的靶向性药物缀合物,其中所述抗体片段是单域抗体。
[0490]
83.根据实施方案82的靶向性药物缀合物,其中所述单域抗体是骆驼(camelid)vhh抗体片段或人源化骆驼vhh抗体片段。
[0491]
84.根据实施方案1至62中任一项的靶向性药物缀合物,其中所述靶向部分是非基于免疫球蛋白的。
[0492]
85.根据实施方案1至84中任一项的靶向性药物缀合物,其中所述细胞表面分子是人细胞表面分子。
[0493]
86.根据实施方案1至85中任一项的靶向性药物缀合物,其中所述细胞表面分子是fap、pdgfr-β、fgfr1、ppar-γ、fsp1、gfap、肌成束蛋白(fascin)、cd147、cxcr4、αvβ6、axl或mertk。
[0494]
87.根据实施方案86的靶向性药物缀合物,其中所述细胞表面分子是fap。
[0495]
88.根据实施方案87的靶向性药物缀合物,其中相对于可溶性fap,所述靶向部分优先与膜结合性fap结合。
[0496]
89.根据实施方案86的靶向性药物缀合物,其中所述细胞表面分子是pdgfr-β。
[0497]
90.根据实施方案86的靶向性药物缀合物,其中所述细胞表面分子是fgfr1。
[0498]
91.根据实施方案86的靶向性药物缀合物,其中所述细胞表面分子是ppar-γ。
[0499]
92.根据实施方案86的靶向性药物缀合物,其中所述细胞表面分子是fsp1。
[0500]
93.根据实施方案86的靶向性药物缀合物,其中所述细胞表面分子是gfap。
[0501]
94.根据实施方案86的靶向性药物缀合物,其中所述细胞表面分子是肌成束蛋白。
[0502]
95.根据实施方案86的靶向性药物缀合物,其中所述细胞表面分子是cd147。
[0503]
96.根据实施方案86的靶向性药物缀合物,其中所述细胞表面分子是cxcr4。
[0504]
97.根据实施方案86的靶向性药物缀合物,其中所述细胞表面分子是αvβ6。
[0505]
98.根据实施方案86的靶向性药物缀合物,其中所述细胞表面分子是axl。
[0506]
99.根据实施方案86的靶向性药物缀合物,其中所述细胞表面分子是mertk。
[0507]
100.根据实施方案1至85中任一项的靶向性药物缀合物,其中所述细胞表面分子是lrrc15。
[0508]
101.根据实施方案1至100中任一项的靶向性药物缀合物,其促进与所述靶向性药物缀合物接触的肌成纤维细胞的凋亡。
[0509]
102.根据实施方案1至100中任一项的靶向性药物缀合物,其促进与所述靶向性药物缀合物接触的肌成纤维细胞的去分化。
[0510]
103.根据实施方案102的靶向性药物缀合物,其中通过平滑肌肌动蛋白表达的减少来测量去分化。
[0511]
104.根据实施方案1至103中任一项的靶向性药物缀合物,其包含具有一个或多个减少效应子功能的氨基酸取代的fc域。
[0512]
105.根据实施方案104的靶向性药物缀合物,其中所述一个或多个取代包含n297a、n297q、n297g、d265a/n297a、d265a/n297g、l235e、l234a/l235a、l234a/l235a/p329a、l234d/l235e:l234r/l235r/e233k、l234d/l235e/d265s:e233k/l234r/l235r/d265s、l234d/l235e/e269k:e233k/l234r/l235r/e269k、l234d/l235e/k322a:e233k/l234r/l235r/k322a、l234d/l235e/p329w:e233k/l234r/l235r/p329w、l234d/l235e/e269k/d265s/k322a:e233k/l234r/l235r/e269k/d265s/k322a或l234d/l235e/e269k/d265s/k322e/e333k:e233k/l234r/l235r/e269k/d265s/k322e/e333k。
[0513]
106.根据实施方案105的靶向性药物缀合物,其中所述一个或多个取代包含n297a。
[0514]
107.根据实施方案105的靶向性药物缀合物,其中所述一个或多个取代包含n297q。
[0515]
108.根据实施方案105的靶向性药物缀合物,其中所述一个或多个取代包含n297g。
[0516]
109.根据实施方案105的靶向性药物缀合物,其中所述一个或多个取代包含d265a/n297a。
[0517]
110.根据实施方案105的靶向性药物缀合物,其中所述一个或多个取代包含d265a/n297g。
[0518]
111.根据实施方案105的靶向性药物缀合物,其中所述一个或多个取代包含l235e。
[0519]
112.根据实施方案105的靶向性药物缀合物,其中所述一个或多个取代包含l234a/l235a。
[0520]
113.根据实施方案105的靶向性药物缀合物,其中所述一个或多个取代包含l234a/l235a/p329a。
[0521]
114.根据实施方案105的靶向性药物缀合物,其中所述一个或多个取代包含l234d/l235e:l234r/l235r/e233k。
[0522]
115.根据实施方案105的靶向性药物缀合物,其中所述一个或多个取代包含l234d/l235e/d265s:e233k/l234r/l235r/d265s。
[0523]
116.根据实施方案105的靶向性药物缀合物,其中所述一个或多个取代包含l234d/l235e/e269k:e233k/l234r/l235r/e269k。
[0524]
117.根据实施方案105的靶向性药物缀合物,其中所述一个或多个取代包含l234d/l235e/k322a:e233k/l234r/l235r/k322a。
[0525]
118.根据实施方案105的靶向性药物缀合物,其中所述一个或多个取代包含l234d/l235e/p329w:e233k/l234r/l235r/p329w。
[0526]
119.根据实施方案105的靶向性药物缀合物,其中所述一个或多个取代包含l234d/l235e/e269k/d265s/k322a:e233k/l234r/l235r/e269k/d265s/k322a。
[0527]
120.根据实施方案105的靶向性药物缀合物,其中所述一个或多个取代包含l234d/l235e/e269k/d265s/k322e/e333k:e233k/l234r/l235r/e269k/d265s/k322e/e333k。
[0528]
121.药物组合物,其包含根据实施方案1至120中任一项的靶向性药物缀合物和药学上可接受的载体。
[0529]
122.根据实施方案121的药物组合物,其中所述药物组合物中至少30%的所述靶向性药物缀合物分子具有在1至30之间的alk5抑制剂:靶向部分比率。
[0530]
123.根据实施方案121的药物组合物,其中所述药物组合物中至少30%的所述靶向性药物缀合物分子具有在1和20之间的alk5抑制剂:靶向部分比率。
[0531]
124.根据实施方案121的药物组合物,其中所述药物组合物中至少30%的所述靶向性药物缀合物分子具有在1至15之间的alk5抑制剂:靶向部分比率。
[0532]
125.根据实施方案121的药物组合物,其中所述药物组合物中至少30%的所述靶向性药物缀合物分子具有在2至12之间的alk5抑制剂:靶向部分比率。
[0533]
126.根据实施方案121的药物组合物,其中所述药物组合物中至少30%的所述靶向性药物缀合物分子具有在4至15之间的alk5抑制剂:靶向部分比率。
[0534]
127.根据实施方案121的药物组合物,其中所述药物组合物中至少30%的所述靶向性药物缀合物分子具有在6至12之间的alk5抑制剂:靶向部分比率。
[0535]
128.根据实施方案121的药物组合物,其中所述药物组合物中至少30%的所述靶向性药物缀合物分子具有在2至8之间的alk5抑制剂:靶向部分比率。
[0536]
129.根据实施方案121的药物组合物,其中所述药物组合物中至少40%的所述靶向性药物缀合物分子具有在1至30之间的alk5抑制剂:靶向部分比率。
[0537]
130.根据实施方案121的药物组合物,其中所述药物组合物中至少40%的所述靶向性药物缀合物分子具有在1至20之间的alk5抑制剂:靶向部分比率。
[0538]
131.根据实施方案121的药物组合物,其中所述药物组合物中至少40%的所述靶向性药物缀合物分子具有在1至15之间的alk5抑制剂:靶向部分比率。
[0539]
132.根据实施方案121的药物组合物,其中所述药物组合物中至少40%的所述靶向性药物缀合物分子具有在2至12之间的alk5抑制剂:靶向部分比率。
[0540]
133.根据实施方案121的药物组合物,其中所述药物组合物中至少40%的所述靶向性药物缀合物分子具有在4至15之间的alk5抑制剂:靶向部分比率。
[0541]
134.根据实施方案121的药物组合物,其中所述药物组合物中至少40%的所述靶向性药物缀合物分子具有在6至12之间的alk5抑制剂:靶向部分比率。
[0542]
135.根据实施方案121的药物组合物,其中所述药物组合物中至少40%的所述靶向性药物缀合物分子具有在2至8之间的alk5抑制剂:靶向部分比率。
[0543]
136.根据实施方案121的药物组合物,其中所述药物组合物中至少50%的所述靶向性药物缀合物分子具有在1至30之间的alk5抑制剂:靶向部分比率。
[0544]
137.根据实施方案121的药物组合物,其中所述药物组合物中至少50%的所述靶向性药物缀合物分子具有在1至20之间的alk5抑制剂:靶向部分比率。
[0545]
138.根据实施方案121的药物组合物,其中所述药物组合物中至少50%的所述靶向性药物缀合物分子具有在1至15之间的alk5抑制剂:靶向部分比率。
[0546]
139.根据实施方案121的药物组合物,其中所述药物组合物中至少50%的所述靶向性药物缀合物分子具有在2至12之间的alk5抑制剂:靶向部分比率。
[0547]
140.根据实施方案121的药物组合物,其中所述药物组合物中至少50%的所述靶向性药物缀合物分子具有在4至15之间的alk5抑制剂:靶向部分比率。
[0548]
141.根据实施方案121的药物组合物,其中所述药物组合物中至少50%的所述靶向性药物缀合物分子具有在6至12之间的alk5抑制剂:靶向部分比率。
[0549]
142.根据实施方案121的药物组合物,其中所述药物组合物中至少50%的所述靶向性药物缀合物分子具有在2至8之间的alk5抑制剂:靶向部分比率。
[0550]
143.根据实施方案121的药物组合物,其中所述药物组合物中至少60%的所述靶向性药物缀合物分子具有在1至30之间的alk5抑制剂:靶向部分比率。
[0551]
144.根据实施方案121的药物组合物,其中所述药物组合物中至少60%的所述靶向性药物缀合物分子具有在1至20之间的alk5抑制剂:靶向部分比率。
[0552]
145.根据实施方案121的药物组合物,其中所述药物组合物中至少60%的所述靶向性药物缀合物分子具有在1至15之间的alk5抑制剂:靶向部分比率。
[0553]
146.根据实施方案121的药物组合物,其中所述药物组合物中至少60%的所述靶向性药物缀合物分子具有在2至12之间的alk5抑制剂:靶向部分比率。
[0554]
147.根据实施方案121的药物组合物,其中所述药物组合物中至少60%的所述靶向性药物缀合物分子具有在4至15之间的alk5抑制剂:靶向部分比率。
[0555]
148.根据实施方案121的药物组合物,其中所述药物组合物中至少60%的所述靶向性药物缀合物分子具有在6至12之间的alk5抑制剂:靶向部分比率。
[0556]
149.根据实施方案121的药物组合物,其中所述药物组合物中至少60%的所述靶向性药物缀合物分子具有在2至8之间的alk5抑制剂:靶向部分比率。
[0557]
150.在有需要的受试者中治疗纤维化的方法,其包括向所述受试者施用根据实施方案1至120中任一项的靶向性药物缀合物或根据实施方案121至149中任一项的药物组合物。
[0558]
151.根据实施方案150的方法,其中所述纤维化是肺纤维化。
[0559]
152.根据实施方案151的方法,其中所述纤维化是特发性肺纤维化(ipf)。
[0560]
153.根据实施方案150的方法,其中所述纤维化是肝纤维化。
[0561]
154.根据实施方案150的方法,其中所述纤维化是肾纤维化。
[0562]
155.根据实施方案150的方法,其中所述纤维化是心脏纤维化。
[0563]
156.根据实施方案150的方法,其中所述纤维化是皮肤纤维化。
[0564]
157.根据实施方案150的方法,其中所述纤维化是食道纤维化。
[0565]
158.根据实施方案153的方法,其中所述受试者患有nash,例如已诊断患有nash。
[0566]
159.根据实施方案150至157中任一项的方法,其中所述受试者患有系统性硬化症,例如已被诊断患有系统性硬化症。
[0567]
160.治疗患有,例如已被诊断患有系统性硬化症的受试者的方法,其包括向所述受试者施用根据实施方案1至120中任一项的靶向性药物缀合物或根据实施方案121至149
中任一项的药物组合物。
[0568]
161.治疗患有nash的受试者,例如已被诊断患有nash的受试者的方法,其包括向所述受试者施用根据实施方案1至120中任一项的靶向性药物缀合物或根据实施方案121至149中任一项的药物组合物。
[0569]
162.根据权利要求160或实施方案161的方法,其中所述受试者表现出纤维化的体征和/或症状。
[0570]
163.根据实施方案160或实施方案161的方法,其中所述受试者不表现出纤维化的体征和/或症状。
[0571]
164.根据实施方案150至163中任一项的方法,其中所述靶向性药物缀合物或药物组合物作为组合疗法方案的一部分施用,该组合疗法方案包括施用一种或多种第二治疗剂,任选地其中所述一种或多种药剂不是根据实施方案1至120中任一项的靶向性药物缀合物(各自为“第二治疗剂”)。
[0572]
165.根据实施方案164的方法,其中所述靶向性药物缀合物或药物组合物与标准护理疗法或治疗方案组合施用。
[0573]
166.根据实施方案164或165的方法,其中所述组合疗法包括向所述受试者施用至少一种第二治疗剂。
[0574]
167.根据实施方案164至166中任一项的方法,其中第二治疗剂包含吡非尼酮(pirfenidone)、尼达尼布(nintedanib)、pentraxin-2、pamrevlumab、泼尼松(prednisone)、可的松(cortisone)、环磷酰胺(cyclophosphamide)或硫唑嘌呤(azathioprine)。
[0575]
168.根据实施方案167的方法,其中第二治疗剂包含吡非尼酮。
[0576]
169.根据实施方案167或实施方案168的方法,其中第二治疗剂包含尼达尼布。
[0577]
170.根据实施方案167至169中任一项的方法,其中第二治疗剂包含pentraxin-2。
[0578]
171.根据实施方案167至170中任一项的方法,其中第二治疗剂包含pamrevlumab。
[0579]
172.根据实施方案167至171中任一项的方法,其中第二治疗剂包含泼尼松。
[0580]
173.根据实施方案167至172中任一项的方法,其中第二治疗剂包含可的松。
[0581]
174.根据实施方案167至173中任一项的方法,其中第二治疗剂包含环磷酰胺。
[0582]
175.根据实施方案167至174中任一项的方法,其中第二治疗剂包含硫唑嘌呤。
[0583]
176.根据实施方案164至175中任一项的方法,其包括用所述组合疗法治疗所述受试者。
[0584]
177.根据实施方案164至176中任一项的方法,其包括向所述受试者施用所述第二治疗剂。
[0585]
178.治疗患有癌症的受试者的方法,其包括向有需要的受试者施用根据实施方案1至120中任一项的靶向性药物缀合物或根据实施方案121至149中任一项的药物组合物。
[0586]
179.根据实施方案178的方法,其中所述癌症是尿路上皮癌(urothelial cancer)。
[0587]
180.根据实施方案179的方法,其中所述癌症是膀胱癌。
[0588]
181.根据实施方案179的方法,其中所述癌症是尿道癌。
[0589]
182.根据实施方案179的方法,其中所述癌症是输尿管癌。
[0590]
183.根据实施方案178的方法,其中所述癌症是肺癌。
[0591]
184.根据实施方案183的方法,其中所述癌症是nsclc。
[0592]
185.根据实施方案184的方法,其中所述nsclc是腺癌。
[0593]
186.根据实施方案184的方法,其中所述nsclc是鳞状细胞癌。
[0594]
187.根据实施方案184的方法,其中所述nsclc是大细胞癌。
[0595]
188.根据实施方案183的方法,其中所述癌症是小细胞肺癌。
[0596]
189.根据实施方案178的方法,其中所述癌症是乳腺癌。
[0597]
190.根据实施方案178的方法,其中所述癌症是胰腺癌。
[0598]
191.根据实施方案178的方法,其中所述癌症是前列腺癌。
[0599]
192.根据实施方案178的方法,其中所述癌症是食道癌。
[0600]
193.根据实施方案178的方法,其中所述癌症是结直肠癌。
[0601]
194.根据实施方案193的方法,其中所述结直肠癌是腺癌。
[0602]
195.根据实施方案193的方法,其中所述结直肠癌是类癌瘤。
[0603]
196.根据实施方案193的方法,其中所述结直肠癌是胃肠道间质瘤。
[0604]
197.根据实施方案193的方法,其中所述结直肠癌是结直肠淋巴瘤。
[0605]
198.根据实施方案178的方法,其中所述癌症是头颈癌。
[0606]
199.根据实施方案178的方法,其中所述癌症是卵巢癌。
[0607]
200.根据实施方案178的方法,其中所述癌症是肾脏癌。
[0608]
201.根据实施方案178的方法,其中所述癌症是胃腺癌。
[0609]
202.根据实施方案178至201中任一项的方法,其中所述靶向性药物缀合物或药物组合物作为组合疗法方案的一部分施用,该组合疗法方案包括施用一种或多种第二治疗剂,任选地其中所述一种或多种药剂不是根据实施方案1至120中任一项的靶向性药物缀合物(各自为“第二治疗剂”)。
[0610]
203.根据实施方案202的方法,其中所述靶向性药物缀合物或药物组合物与标准护理疗法或治疗方案组合施用。
[0611]
204.根据实施方案202或203的方法,其中所述组合疗法包括向所述受试者施用至少一种第二治疗剂。
[0612]
205.根据实施方案202至204中任一项的方法,其中所述组合疗法包含免疫疗法,任选地其中所述免疫疗法是检查点抑制剂疗法、嵌合抗原受体(car)疗法、过继性t细胞疗法、溶瘤病毒疗法、树突细胞疫苗疗法、sting激动剂疗法、tlr激动剂疗法、瘤内cpg疗法或细胞因子疗法。
[0613]
206.根据实施方案202至205中任一项的方法,其中所述组合疗法包含检查点抑制剂疗法。
[0614]
207.根据实施方案206的方法,其中所述检查点抑制剂疗法包含t细胞检查点抑制剂疗法。
[0615]
208.根据实施方案207的方法,其中所述t细胞检查点抑制剂疗法包含抗体或其抗原结合片段。
[0616]
209.根据实施方案206至208中任一项的方法,其中所述检查点抑制剂疗法靶向pd1、pdl1、ctla4、tigit、lag3、ox40、cd40 vista或其组合。
[0617]
210.根据实施方案209的方法,其中所述检查点抑制剂疗法靶向pd1。
[0618]
211.根据实施方案210的方法,其中第二治疗剂是派姆单抗。
[0619]
212.根据实施方案210的方法,其中第二治疗剂是纳武单抗。
[0620]
213.根据实施方案210的方法,其中第二治疗剂是西米普利单抗。
[0621]
214.根据实施方案210的方法,其中第二治疗剂是多塔利单抗。
[0622]
215.根据实施方案209至214中任一项的方法,其中检查点抑制剂疗法靶向pdl1。
[0623]
216.根据实施方案215的方法,其中第二治疗是阿特珠单抗。
[0624]
217.根据实施方案215的方法,其中第二治疗剂是阿维单抗。
[0625]
218.根据实施方案215的方法,其中第二治疗剂是德瓦鲁单抗。
[0626]
219.根据实施方案209至218中任一项的方法,其中所述检查点抑制剂疗法靶向ctla4。
[0627]
220.根据实施方案219的方法,其中第二治疗剂是伊匹单抗。
[0628]
221.根据实施方案209至220中任一项的方法,其中所述检查点抑制剂疗法靶向tigit。
[0629]
222.根据实施方案221的方法,其中第二治疗剂是etigilimab。
[0630]
223.根据实施方案221的方法,其中第二治疗剂是tiragolumab。
[0631]
224.根据实施方案221的方法,其中第二治疗剂是ab154。
[0632]
225.根据实施方案209至224中任一项的方法,其中检查点抑制剂疗法靶向lag3。
[0633]
226.根据实施方案225的方法,其中第二治疗剂是lag525。
[0634]
227.根据实施方案225的方法,其中第二治疗剂是sym022。
[0635]
228.根据实施方案225的方法,其中第二治疗剂是relatlimab。
[0636]
229.根据实施方案225的方法,其中第二治疗剂是tsr-033。
[0637]
230.根据实施方案209至229中任一项的方法,其中所述检查点抑制剂疗法靶向ox40。
[0638]
231.根据实施方案230的方法,其中第二治疗剂是medi6469。
[0639]
232.根据实施方案230的方法,其中第二治疗剂是pf-04518600。
[0640]
233.根据实施方案230的方法,其中第二治疗剂是bms 986178。
[0641]
234.根据实施方案209至233中任一项的方法,其中检查点抑制剂疗法靶向cd40。
[0642]
235.根据实施方案234的方法,其中第二治疗剂是selicrelumab。
[0643]
236.根据实施方案234的方法,其中第二治疗剂是cp-870,893。
[0644]
237.根据实施方案234的方法,其中第二治疗剂是apx005m。
[0645]
238.根据实施方案209至237中任一项的方法,其中所述检查点抑制剂疗法靶向vista。
[0646]
239.根据实施方案238的方法,其中第二治疗剂是hmbd-002。
[0647]
240.根据实施方案202至239中任一项的方法,其中第二治疗剂是嵌合抗原受体(car)。
[0648]
241.根据实施方案202至240中任一项的方法,其中所述组合疗法包含过继性t细胞疗法。
[0649]
242.根据实施方案241的方法,其中所述过继性t细胞疗法是自体t细胞疗法。
[0650]
243.根据实施方案202至242中任一项的方法,其中所述组合疗法包含溶瘤病毒疗法。
[0651]
244.根据实施方案202至243中任一项的方法,其中所述组合疗法包含树突细胞疫苗疗法。
[0652]
245.根据实施方案202至244中任一项的方法,其中所述组合疗法包含sting激动剂疗法。
[0653]
246.根据实施方案202至245中任一项的方法,其中所述组合疗法包含tlr激动剂疗法。
[0654]
247.根据实施方案202至246中任一项的方法,其中所述组合疗法包含化疗。
[0655]
248.根据实施方案247的方法,其中第二治疗剂是抗代谢物、烷化剂、蒽环类药物、抗微管剂、铂化合物、紫杉烷、拓扑异构酶抑制剂或长春花生物碱。
[0656]
249.根据实施方案248的方法,其中第二治疗剂是抗代谢物。
[0657]
250.根据实施方案249的方法,其中所述抗代谢物是5-氟尿嘧啶(5-fluorouracil)。
[0658]
251.根据实施方案249的方法,其中所述抗代谢物是吉西他滨(gemcitabine)。
[0659]
252.根据实施方案249的方法,其中所述抗代谢物是甲氨蝶呤(methotrexate)。
[0660]
253.根据实施方案248的方法,其中第二治疗剂是烷化剂。
[0661]
254.根据实施方案253的方法,其中所述烷化剂是环磷酰胺(cyclophosphamide)。
[0662]
255.根据实施方案253的方法,其中所述烷化剂是达卡巴嗪(dacarbazine)。
[0663]
256.根据实施方案253的方法,其中烷基化剂是氮芥(mechlorethamine)。
[0664]
257.根据实施方案253的方法,其中所述烷化剂是地吖醌(diaziquone)。
[0665]
258.根据实施方案253的方法,其中所述烷化剂是替莫唑胺(temozolomide)。
[0666]
259.根据实施方案248的方法,其中第二治疗剂是蒽环类药物。
[0667]
260.根据实施方案259的方法,其中所述蒽环类药物是多柔比星(doxorubicin)。
[0668]
261.根据实施方案259的方法,其中所述蒽环类是表柔比星(epirubicin)。
[0669]
262.根据实施方案248的方法,其中第二治疗剂是抗微管剂。
[0670]
263.根据实施方案262的方法,其中所述抗微管剂是长春碱(vinblastine)。
[0671]
264.根据实施方案248的方法,其中第二治疗剂是铂化合物。
[0672]
265.根据实施方案264的方法,其中所述铂化合物是顺铂(cisplatin)。
[0673]
266.根据实施方案264的方法,其中所述铂化合物是奥沙利铂(oxaliplatin)。
[0674]
267.根据实施方案248的方法,其中第二治疗剂是紫杉烷。
[0675]
268.根据实施方案267的方法,其中所述紫杉烷是紫杉醇(paclitaxel)。
[0676]
269.根据实施方案267的方法,其中所述紫杉烷是多西紫杉醇(docetaxel)。
[0677]
270.根据实施方案248的方法,其中第二治疗剂是拓扑异构酶抑制剂。
[0678]
271.根据实施方案270的方法,其中所述拓扑异构酶抑制剂是依托泊苷(etoposide)。
[0679]
272.根据实施方案270的方法,其中拓扑异构酶抑制剂是米托蒽醌(mitoxantrone)。
[0680]
273.根据实施方案248的方法,其中第二治疗剂是长春花生物碱。
[0681]
274.根据实施方案273的方法,其中所述长春花生物碱是长春新碱(vincristine)。
[0682]
275.根据实施方案202至274中任一项的方法,其中所述组合疗法包含瘤内cpg疗法。
[0683]
276.根据实施方案202至275中任一项的方法,其中第二治疗剂是具有细胞毒性有效载荷(payload)的adc。
[0684]
277.根据实施方案276的方法,其中所述具有细胞毒性有效载荷的adc靶向fap。
[0685]
278.根据实施方案277的方法,其中第二治疗剂是omtx705。
[0686]
279.根据实施方案202至278中任一项的方法,其中第二治疗剂是细胞因子。
[0687]
280.根据实施方案279的方法,其中所述细胞因子是il2。
[0688]
281.根据实施方案279的方法,其中细胞因子是il12。
[0689]
282.根据实施方案279的方法,其中所述细胞因子是ifn-α。
[0690]
283.根据实施方案279的方法,其中所述细胞因子是ifn-γ。
[0691]
284.根据实施方案202至283中任一项的方法,其包括用所述组合疗法治疗所述受试者。
[0692]
285.根据实施方案202至284中任一项的方法,其包括向所述受试者施用所述第二治疗剂。
[0693]
286.促进肌成纤维细胞去分化为静止成纤维细胞的方法,其包括使该肌成纤维细胞与根据实施方案1至120中任一项的靶向性药物缀合物或根据实施方案121至149中任一项的药物组合物接触。
[0694]
287.促进活化的成纤维细胞去分化为静止(resting)成纤维细胞的方法,其包括使该活化的成纤维细胞与根据实施方案1至120中任一项的靶向性药物缀合物或根据实施方案121至149中任一项的药物组合物接触。
[0695]
288.促进向肌成纤维细胞转变的成纤维细胞去分化为静止成纤维细胞的方法,其包括使所述向肌成纤维细胞转变的成纤维细胞与根据实施方案1至120中任一项的靶向性药物缀合物或根据实施方案121至149中任一项的药物组合物接触。
[0696]
289.根据实施方案286至288中任一项的方法,其中去分化包括平滑肌肌动蛋白表达的减少。
[0697]
290.促进肌成纤维细胞凋亡的方法,其包括使所述肌成纤维细胞与根据实施方案1至120中任一项的靶向性药物缀合物或根据实施方案121至149中任一项的药物组合物接触。
[0698]
291.促进活化的成纤维细胞凋亡的方法,其包括使所述活化的成纤维细胞与根据实施方案1至120中任一项的靶向性药物缀合物或根据实施方案121至149中任一项的药物组合物接触。
[0699]
292.促进向肌成纤维细胞转变的成纤维细胞凋亡的方法,其包括使所述向肌成纤维细胞转化的成纤维细胞与根据实施方案1至120中任一项的靶向性药物缀合物或根据实施方案121至149中任一项的药物组合物接触。
[0700]
293.根据实施方案286至292中任一项的方法,其中所述接触在受试者体内进行。
[0701]
294.根据权利要求293的方法,其包括向所述受试者施用所述靶向性药物缀合物
或药物组合物。
[0702]
295.化合物c或其盐。
[0703]
296.化合物n或其盐。
[0704]
297.靶向性药物缀合物,其包含与靶向部分可操作地连接的化合物c。
[0705]
298.靶向性药物缀合物,其包含与靶向部分可操作地连接的化合物n。
[0706]
299.与接头缀合的化合物c。
[0707]
300.与接头缀合的化合物n。
[0708]
虽然已经阐释和描述了各种具体实施方案,但是应当理解,可以在不背离本公开的精神和范围的情况下实施各种改变。
[0709]
8.参考文献引用
[0710]
本技术中引用的所有出版物、专利、专利申请和其他文件均通过引用整体并入本文以用于所有目的,就如各个个体的出版物、专利、专利申请或其他文件单独表明以引用方式并入以用于所有目的。如果在本文并入的一个或多个参考文献的教导与本公开之间存在不一致的情况,则意旨本说明书的教导。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。