1.本发明属于成分检测领域,具体涉及一种七清败毒颗粒的指纹图谱的构建方法和其成分含量的测定方法及应用。
背景技术:
2.七清败毒颗粒具有清热解毒,燥湿止泻的功效,主治湿热泄泻,雉鸡白痢。七清败毒颗粒处方中含有黄芩、虎杖、白头翁、苦参、板蓝根、绵马贯众、大青叶、蔗糖和糊精等中药成分。其药效、安全性与组分及其含量相关,但因其成分复杂,各成分的含量不易控制,导致成品的质量控制相对困难。
3.《兽药典》(2010年二部)公开了一种使用薄层色谱法对七清败毒颗粒中的黄芩苷、虎杖、大黄素、靛蓝和靛玉红进行成分鉴别,步骤繁琐,且没有对其各成分进行定量。因此该方法对于七清败毒颗粒质量控制没有达到量化的标准,也无法对其中的其它组分进行定性定量。因此,目前亟需高效、能同时定性定量的七清败毒颗粒的检测方法,为其质量控制提供技术支持。
技术实现要素:
4.针对上述现有技术提及的当中七清败毒颗粒的检测步骤繁琐、无法同时对多种成分定性定量的问题,本发明提供一种七清败毒颗粒的指纹图谱的构建方法和其成分含量的测定方法及应用,特别涉及检测七清败毒颗粒中的(r,s)-告依春、黄芩苷、黄芩素、汉黄芩素、白头翁皂苷b4、虎杖苷、大黄素,这7种成分的定性定量。
5.本发明采用高效液相色谱(hplc)方法制作七清败毒颗粒的指纹图谱,并对七清败毒颗粒成分含量进行分析,为其质量控制提供参考。
6.为实现上述目的,具体包括以下技术方案:
7.一种七清败毒颗粒的指纹图谱的构建方法,包括如下步骤,制备七清败毒颗粒供试品溶液,将七清败毒颗粒供试品溶液进行高效液相色谱分析,获得七清败毒颗粒的指纹图谱;高效液相色谱分析的色谱条件包括:c-18色谱柱为固定相,以乙腈为流动相a相,0.05-0.2%磷酸水溶液为流动相b。
8.高效液相色谱分析的色谱条件还包括:c-18色谱柱为固定相,以乙腈为流动相a相,0.05-0.2%磷酸水溶液为流动相b,检测波长分别为201nm、245nm,梯度淋洗时,流动相a相和流动相b相的体积分数之和为100%,其中a相的体积分数在0-150min内变化如下:0-25min,2%
→
5%a相;25-45min,5%
→
15%a相;45-70min,15%a相;70-80min,15%
→
25%a相;80-100min,25%a相;100-105min,25%
→
30%a相;105-110min,30%a相;110-120min,30%
→
40%a相;120-130min,40%
→
45%a相;130-140min,45%a相;140-150min,45%
→
80%a相。
9.本发明的发明人在大量的七清败毒颗粒指纹图谱研究中发现,通过以乙腈为流动
70min,15%a相;70-80min,15%
→
25%a相;80-100min,25%a相;100-105min,25%
→
30%a相;105-110min,30%a相;110-120min,30%
→
40%a相;120-130min,40%
→
45%a相;130-140min,45%a相;140-150min,45%
→
80%a相。
23.所述的高效液相色谱分析的色谱条件还包括:柱温为25-35℃,进样量为5-15μl,流动相流速为0.5-1.5ml/min。
24.进一步优选地,0.1%磷酸水溶液为流动相b,柱温为30℃,进样量为10μl,流速为1ml/min,此时得到的色谱图具有更好的基线稳定性、分离度、峰型对称性。
25.优选地,所述的外标法通过计算高效液相色谱的谱峰面积,并将其作为纵坐标(y),以其成分的浓度为横坐标(x),建立每种成分的线性回归方程。
26.本发明的方法得到每种成分的线性回归方程的r值非常接近1或等于1,线性好,为成分的定量提供可靠的依据。
27.所述的外标法中,包括对照品溶液,所述的对照品溶液为以下a-e中任一项的溶液:
28.a:对照药材溶液;
29.b:阴性样品溶液;
30.c:混合对照品溶液;
31.d:混合对照品溶液和对照药材溶液;
32.e:混合对照品溶液和阴性样品溶液。
33.通过分析供试品溶液和对照药材溶液的hplc谱图,能够得到七清败毒颗粒的中药成分信息。
34.通过分析供试品溶液和阴性样品溶液的hplc谱图,能够得到七清败毒颗粒的中药成分信息。
35.通过分析供试品溶液和混合对照品溶液的hplc谱图,能够得到七清败毒颗粒的化学成分信息。
36.通过分析供试品溶液、混合对照品溶液和对照药材溶液的hplc谱图,能够得到七清败毒颗粒的中药成分信息、化学成分及其含量信息。
37.通过分析供试品溶液、混合对照品溶液和阴性样品溶液的hplc谱图,能够得到七清败毒颗粒的中药成分信息、化学成分及其含量信息。
38.所述的中药成分包括如下成分中的至少一种:黄芩、虎杖、白头翁、苦参、板蓝根、绵马贯众、大青叶。
39.所述的化学成分包括如下成分中的至少一种:(r,s)-告依春、虎杖苷,黄芩苷,白头翁皂苷b4、黄芩素,汉黄芩素、大黄素。
40.优选地,供试品溶液的制备方法包括如下步骤:将七清败毒颗粒与溶剂混合,经过超声提取或回流提取后,得到供试品溶液;所述的溶剂为醇或醇水混合溶液。
41.优选地,对照药材溶液的制备方法包括如下步骤:将药材与溶剂混合,经过超声提取或回流提取后,得到对照药材溶液;所述的溶剂为醇或醇水混合溶液。
42.优选地,所述的药材的浓度为2-15g/l。
43.所述的药材为黄芩、虎杖、白头翁、苦参、板蓝根、绵马贯众、大青叶中的一种。
44.所述的对照药材溶液为以下溶液中的一种:黄芩对照药材溶液、白头翁对照药材
溶液、虎杖对照药材溶液、苦参对照药材溶液、板蓝根对照药材溶液、大青叶对照药材溶液、绵马贯众对照药材溶液。
45.优选地,阴性样品溶液的制备方法包括如下步骤:将阴性样品与溶剂混合,经过超声提取或回流提取后,得到阴性样品溶液;所述的阴性样品为缺少以下任一种组分后剩余的样品:黄芩、虎杖、白头翁、苦参、板蓝根、绵马贯众、大青叶;所述的溶剂为醇或醇水混合溶液。
46.所述的阴性样品溶液包括以下溶液:黄芩阴性样品溶液、白头翁阴性样品溶液、虎杖阴性样品溶液、苦参阴性样品溶液、板蓝根阴性样品溶液、大青叶阴性样品溶液、绵马贯众阴性样品溶液。
47.阴性样品中黄芩、虎杖、白头翁、苦参、板蓝根、绵马贯众、大青叶的质量比为(1-4):(1-2):(0.5-1):(1-2):(1-3):(0.5-1):(1-2),其中缺少的任一组分质量视为0。
48.进一步优选地,黄芩、虎杖、白头翁、苦参、板蓝根、绵马贯众、大青叶的质量比为4:1.3:0.875:1.75:2.66:0.87:1.75,其中缺少的任一组分质量视为0。
49.优选地,所述的混合对照品溶液的制备方法包括如下步骤:将(r,s)-告依春、虎杖苷,黄芩苷,白头翁皂苷b4、黄芩素、汉黄芩素和大黄素溶解于溶剂中,得到混合对照品溶液;所述的溶剂为醇或醇水混合溶液。
50.进一步优选地,所述的混合对照品溶液中各组分及其浓度如下:0.0003-0.015mg/ml(r,s)-告依春,0.008-0.4mg/ml虎杖苷,0.08-4mg/ml黄芩苷,0.02-1mg/ml白头翁皂苷b4,0.01-0.5mg/ml黄芩素,0.004-0.2mg/ml汉黄芩素,0.0015-0.075mg/ml大黄素。
51.更进一步优选地,所述的混合对照品溶液中各组分及其浓度如下:0.003mg/ml(r,s)-告依春,0.08mg/ml虎杖苷,0.8mg/ml黄芩苷,0.2mg/ml白头翁皂苷b4,0.1mg/ml黄芩素,0.04mg/ml汉黄芩素,0.015mg/ml大黄素。
52.优选地,所述的超声提取的时间为20-90min。
53.进一步优选地,所述的超声提取的时间为30min。
54.优选地,所述的超声提取的时间为室温。
55.所述的回流提取的时间为20-90min。
56.进一步优选地,所述的回流提取的时间为30min。
57.进一步优选地,所述的醇为甲醇。
58.所述的七清败毒颗粒的指纹图谱的构建方法或七清败毒颗粒的成分含量的检测方法在七清败毒颗粒质量控制方面的应用。
59.七清败毒颗粒的指纹图谱的构建方法或检测方法能够确定其中药成分信息、化学成分信息,特别是关于(r,s)-告依春、虎杖苷,黄芩苷,白头翁皂苷b4、黄芩素,汉黄芩素、大黄素7种化学成分的定性定量,应用在七清败毒颗粒质量控制方面具有较好的前景。
60.相对于现有技术,本发明具有以下优势:
61.(1)本发明的建立了七清败毒颗粒指纹图谱,为质量控制提供参考。
62.(2)本发明的方法能高效、准确地对七清败毒颗粒中的(r,s)-告依春、虎杖苷,黄芩苷,白头翁皂苷b4、黄芩素,汉黄芩素、大黄素进行定性定量。
63.(3)本发明的方法能高效地确定七清败毒颗粒中的中药成分信息,特别是黄芩、虎杖、白头翁、苦参、板蓝根、绵马贯众、大青叶,这7种中药成分。
64.(4)利用本发明的方法得到的色谱图的基线稳定,谱峰分离度好、峰型对称、专属性强。
65.(5)通过本发明的方法,能够方便快捷地检测和鉴定七清败毒颗粒的质量,为其在质量的控制方面提供可靠的、量化的标准。
附图说明
66.图1为245nm波长下供试品溶液、各对照药材溶液和混合对照品溶液hplc图谱,其中,s1-s9分别代表供试品溶液、黄芩对照药材溶液、白头翁对照药材溶液、虎杖对照药材溶液、苦参对照药材溶液、板蓝根对照药材溶液、大青叶对照药材溶液、绵马贯众对照药材溶液、混合对照品溶液的hplc图谱。
67.图2为201nm波长下供试品溶液、各对照药材溶液和混合对照品溶液hplc图谱,其中,s1-s9分别代表供试品溶液、黄芩对照药材溶液、白头翁对照药材溶液、虎杖对照药材溶液、苦参对照药材溶液、板蓝根对照药材溶液、大青叶对照药材溶液、绵马贯众对照药材溶液、混合对照品溶液的hplc图谱。
68.图3为245nm波长下供试品溶液、各阴性样品溶液和混合对照品溶液hplc图谱,其中,s1-s9分别代表供试品溶液、黄芩阴性样品溶液、白头翁阴性样品溶液、虎杖阴性样品溶液、苦参阴性样品溶液、板蓝根阴性样品溶液、大青叶阴性样品溶液、绵马贯众阴性样品溶液、混合对照品溶液的hplc图谱。
69.图4为201nm波长下供试品溶液、各阴性样品溶液和混合对照品溶液hplc图谱,其中,s1-s9分别代表供试品溶液、黄芩阴性样品溶液、白头翁阴性样品溶液、虎杖阴性样品溶液、苦参阴性样品溶液、板蓝根阴性样品溶液、大青叶阴性样品溶液、绵马贯众阴性样品溶液、混合对照品溶液的hplc图谱。
70.图5为245nm波长下自制七清败毒颗粒样品溶液和5种批次的七清败毒颗粒样品溶液hplc图谱,其中,s1代表自制七清败毒颗粒样品溶液,s2-s6分别代表批次2102001、批次2103001、批次2104001、批次2105001、批次2106001的hplc图谱。
71.图6为201nm波长下自制七清败毒颗粒样品溶液和5种批次的七清败毒颗粒样品溶液hplc图谱,其中,s1代表自制七清败毒颗粒样品溶液,s2-s6分别代表批次2102001、批次2103001、批次2104001、批次2105001、批次2106001的hplc图谱。
具体实施方式
72.为更好地说明本发明的目的、技术方案和优点,下面将通过具体实施例对本发明作进一步说明。
73.材料、仪器及测试方法
74.七清败毒颗粒(广东容大生物股份有限公司,批号2006007),黄芩中药配方颗粒(江阴天江药业有限公司,批号20081721),虎杖中药配方颗粒(江阴天江药业有限公司,批号20120501),白头翁中药配方颗粒(江阴天江药业有限公司,批号19091631),苦参中药配方颗粒(江阴天江药业有限公司,批号20071721),板蓝根中药配方颗粒(江阴天江药业有限公司,批号20040641),绵马贯众中药配方颗粒(江阴天江药业有限公司,批号20026934),大青叶中药配方颗粒(江阴天江药业有限公司);(r,s)-告依春(成都曼斯特生物科技有限公
司),虎杖苷(中国药品生物制品检定所),黄芩苷(中国食品药品检定研究院),白头翁皂苷b4(成都曼斯特生物科技有限公司),黄芩素(中国药品生物制品检定所),汉黄芩素(成都曼斯特生物科技有限公司),大黄素(中国食品药品检定研究院);高效液相色谱仪(安捷伦1260infinityⅱ),检测器(vwd,agilent g7114a),色谱柱(phenomenex c-18)。
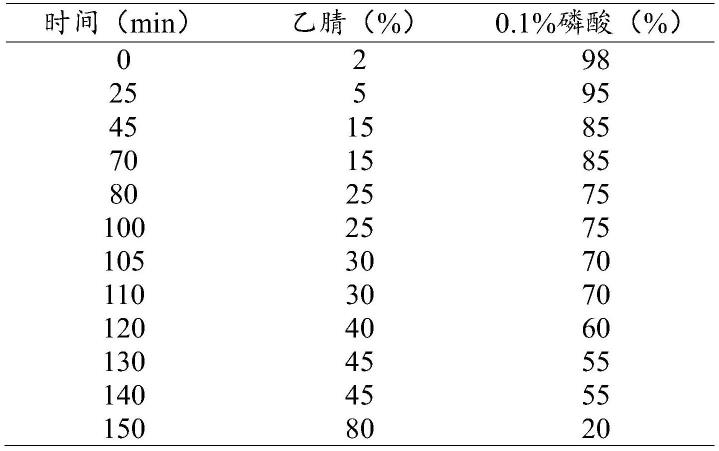
75.以c-18色谱柱为固定相,乙腈为流动相的a相,0.05-0.2%磷酸水溶液为流动相b,柱温为25-35℃,进样量为5-15μl,流动相流速为0.5-1.5ml/min,检测波长分别为201nm、245nm,进行梯度洗脱,流动相梯度程序如下表1所示:
76.表1流动相梯度
[0077][0078]
实施例1
[0079]
1.混合对照品溶液的制备
[0080]
精确称取(r,s)-告依春、虎杖苷,黄芩苷,白头翁皂苷b4、黄芩素、汉黄芩素、大黄素,用甲醇稀释得到6种混合对照品溶液(每种混合对照品溶液中含有7种组分),编号1-6。1-6号中具体的组分及含量分别如下:(r,s)-告依春的浓度分别为0.0003、0.0006、0.0015、0.003、0.006、0.015mg/ml,虎杖苷的浓度分别为0.008、0.016、0.04、0.08、0.16、0.4mg/ml,黄芩苷的浓度分别为0.08、0.16、0.4、0.8、1.6、4mg/ml,白头翁皂苷b4的浓度分别为0.02、0.04、0.1、0.2、0.4、1mg/ml,黄芩素的浓度分别为0.01、0.02、0.05、0.1、0.2、0.5mg/ml,汉黄芩素的浓度分别为0.004、0.008、0.02、0.04、0.08、0.2mg/ml,大黄素的浓度分别为0.0015、0.003、0.0075、0.015、0.03、0.075mg/ml。
[0081]
2.对照药材溶液的制备
[0082]
黄芩对照药材溶液:取黄芩中药配方颗粒0.4g(每1g相当于饮片5g),加甲醇40ml,加热回流提取30min,滤过,取滤液并将滤液的溶剂蒸干,加入80%甲醇(20%水和80%甲醇的混合溶液)使溶解,定容至25ml。作为黄芩对照药材溶液。
[0083]
虎杖对照药材溶液:取虎杖中药配方颗粒0.15g(每1g相当于饮片15g),参照黄芩对照药材溶液方法处理,作为虎杖对照药材溶液。
[0084]
白头翁对照药材溶液:取白头翁中药配方颗粒0.085g(每1g相当于饮片20g)参照黄芩对照药材溶液方法处理,作为白头翁对照药材溶液。
[0085]
苦参对照药材溶液:取苦参中药配方颗粒0.12g(每1g相当于饮片10g),参照黄芩
对照药材溶液方法处理,作为苦参对照药材溶液。
[0086]
板蓝根对照药材溶液:取板蓝根中药配方颗粒0.53g(每1g相当于饮片7.5g),参照黄芩对照药材溶液方法处理,作为板蓝根对照药材溶液。
[0087]
绵马贯众对照药材溶液:取绵马贯众中药配方颗粒0.0875g(每1g相当于饮片15g),参照黄芩对照药材溶液方法处理,作为绵马贯众对照药材溶液。
[0088]
大青叶对照药材溶液:取大青叶中药配方颗粒0.53g(每1g相当于饮片5g),参照黄芩对照药材溶液方法处理,作为大青叶对照药材溶液。
[0089]
3.供试品溶液的制备
[0090]
精确称取七清败毒颗粒粉末10g,加入50ml甲醇,称重,超声提取30min,放至室温,称重,加甲醇补足损失重量,滤过,取续滤液30ml,滤液蒸干,残渣中加入80%甲醇溶解,并用80%甲醇定容至25ml,作为供试品溶液。
[0091]
4.阴性样品溶液制备
[0092]
4.1黄芩阴性样品溶液:取大青叶中药配方颗粒0.175g、绵马贯众中药配方颗粒0.087g、虎杖中药配方颗粒0.133g、白头翁中药配方颗粒0.0875g、苦参中药配方颗粒0.175g、板蓝根中药配方颗粒0.266g,加甲醇40ml,加热回流提取30min,滤过,滤液蒸干,残渣加80%甲醇溶解,并用80%甲醇定容至25ml,作为黄芩阴性样品溶液。
[0093]
4.2虎杖阴性样品溶液:取大青叶中药配方颗粒0.175g、绵马贯众中药配方颗粒0.087g、黄芩中药配方颗粒0.4g、白头翁中药配方颗粒0.0875g、苦参中药配方颗粒0.175g、板蓝根中药配方颗粒0.266g,参照黄芩阴性样品溶液制作方法处理,作为虎杖阴性样品溶液。
[0094]
4.3白头翁阴性样品溶液:取大青叶中药配方颗粒0.175g、绵马贯众中药配方颗粒0.087g、虎杖中药配方颗粒0.133g、黄芩中药配方颗粒0.4g、苦参中药配方颗粒0.175g、板蓝根中药配方颗粒0.266g,参照黄芩阴性样品溶液制作方法处理,作为白头翁阴性样品溶液。
[0095]
4.4苦参阴性样品溶液:取大青叶中药配方颗粒0.175g、绵马贯众中药配方颗粒0.087g、虎杖中药配方颗粒0.133g、白头翁中药配方颗粒0.0875g、黄芩中药配方颗粒0.4g、板蓝根中药配方颗粒0.266g,参照黄芩阴性样品溶液制作方法处理,作为苦参阴性样品溶液。
[0096]
4.5板蓝根阴性样品溶液:取大青叶中药配方颗粒0.175g、绵马贯众中药配方颗粒0.087g、虎杖中药配方颗粒0.133g、白头翁中药配方颗粒0.0875g、黄芩中药配方颗粒0.4g、苦参中药配方颗粒0.175g,参照黄芩阴性样品溶液制作方法处理,作为板蓝根阴性样品溶液。
[0097]
4.6大青叶阴性样品溶液:取苦参中药配方颗粒0.175g、绵马贯众中药配方颗粒0.087g、虎杖中药配方颗粒0.133g、白头翁中药配方颗粒0.0875g、黄芩中药配方颗粒0.4g、板蓝根中药配方颗粒0.266g,参照黄芩阴性样品溶液制作方法处理,作为大青叶阴性样品溶液。
[0098]
4.7绵马贯众阴性样品溶液:取大青叶中药配方颗粒0.175g、苦参中药配方颗粒0.175g、虎杖中药配方颗粒0.133g、白头翁中药配方颗粒0.0875g、黄芩中药配方颗粒0.4g、板蓝根中药配方颗粒0.266g,参照黄芩阴性样品溶液制作方法处理,作为绵马贯众阴性样
品溶液。
[0099]
5.色谱条件的优化
[0100]
5.1流动相b的浓度的优化:将“3.”的供试品进行高效液相色谱检测分析,以c-18色谱柱为固定相,乙腈为流动相a相,检测波长分别为201nm、245nm,柱温为30℃,流速为1ml/min,进样量为10μl,梯度洗脱程序如表1所示,流动相b为磷酸水溶液,仅仅改变磷酸浓度(浓度范围0.05-0.2%),考察不同磷酸浓度对色谱图的影响。结果显示,磷酸的浓度分别为0.05%、0.1%和0.2%时,供试品溶液的谱图基线、峰型对称性、分离度、专属性都较好。
[0101]
5.2流动相流速的优化:将“3.”的供试品溶液进行高效液相色谱检测分析,以c-18色谱柱为固定相,乙腈为流动相a相,流动相b为0.1%磷酸水溶液,检测波长分别为201nm、245nm,柱温为30℃,进样量为10μl,梯度洗脱程序如表1所示,仅仅改变色谱的流动相流速(0.5-1.5ml/min),考察不同液体流速对色谱图的影响。结果显示,流速为0.5ml/min、1.0ml/min、1.5ml/min时,供试品溶液的谱图基线、峰型对称性、分离度、专属性都较好。
[0102]
5.3柱温的优化:将“3.”的供试品溶液进行高效液相色谱检测分析,以c-18色谱柱为固定相,乙腈为流动相a相,流动相b为0.1%磷酸水溶液,检测波长分别为201nm、245nm,进样量为10μl,梯度洗脱程序如表1所示,流速为1ml/min,仅仅改变柱温,考察不同柱温(20-35℃)对色谱图的影响。结果显示,当柱温为25℃、30℃、35℃时,供试品溶液的谱图基线、峰型对称性、分离度、专属性都较好。
[0103]
5.4进样量的优化:将“3.”的供试品溶液进行高效液相色谱检测分析,以c-18色谱柱为固定相,乙腈为流动相a相,检测波长分别为201nm、245nm,梯度洗脱程序如表1所示,流动相b为0.1%磷酸水溶液,流速为1ml/min,柱温为30℃,仅仅改变进样量(5-15μl),考察不同进样量对色谱图的影响。结果显示,进样量为5μl、10μl、15μl时,供试品溶液的谱图基线、峰型对称性、分离度、专属性都较好。
[0104]
5.5检测波长的优化:将“3.”的供试品溶液进行高效液相色谱检测分析,以c-18色谱柱为固定相,乙腈为流动相a相,流动相b相为0.1%磷酸水溶液,梯度洗脱程序如表1所示,流速为1ml/min,柱温为30℃,进样量为10μl,仅仅改变色谱检测时的检测波长(190nm-260nm),考察不同检测波长对色谱图的影响。结果表明,检测波长分别为201nm、245nm时,供试品溶液的谱图中同时显示的成分的谱峰数量最多,专属性较强,峰型的分离度较好,若是采用其他波长作为检测波长,存在某些成分的谱峰显示不完全,或分离度不好,或峰型不对称的现象。
[0105]
6.指纹图谱的建立及成分归属
[0106]
液相色谱检测参数如下:c-18色谱柱为固定相,以乙腈为流动相a相,0.1%磷酸水溶液为流动相b,检测波长分别为201nm、245nm,柱温为30℃,流速为1ml/min,流动相梯度如表1所示。使用实施例1中4号混合对照溶液作为本实施例当中的混合对照溶液进行分析,即:混合对照溶液中(r,s)-告依春浓度为0.003mg/ml、虎杖苷浓度为0.08mg/ml、黄芩苷浓度为0.8mg/ml、白头翁皂苷浓度为b4 0.2mg/ml、黄芩素浓度为0.1mg/ml、汉黄芩素浓度为0.04mg/ml、大黄素浓度为0.015mg/ml。
[0107]
采用供试品溶液、阴性样品溶液、各对照药材溶液、混合对照品溶液hplc图谱对比的方法,在相同色谱条件进行检测,以保留时间作为鉴定标准,进行色谱峰的指认,并对共有峰进行初步归属分析,结果见图1~4。由图1~4可知,(1)根据供试品溶液和对照药材溶
液(或供试品溶液和阴性样品溶液)的hplc谱图可知,七清败毒颗粒中含有如下7种中药成分:黄芩、虎杖、白头翁、苦参、板蓝根、绵马贯众、大青叶。(2)根据供试品溶液和混合对照品溶液的hplc谱图可知,七清败毒颗粒中含有(r,s)-告依春、虎杖苷,黄芩苷,白头翁皂苷b4、黄芩素,汉黄芩素、大黄素7种化学成分。(3)通过分析供试品溶液、混合对照品溶液和对照药材溶液(或供试品溶液、混合对照品溶液和阴性样品溶液)的hplc谱图,七清败毒颗粒中包括如下信息:(a)7种成分得到鉴定:在图1当中,1号峰(r,s)-告依春来源于板蓝根,2号峰虎杖苷和15号峰大黄素来源于虎杖,5号峰黄芩苷、12号峰黄芩素和13号峰汉黄芩素来源于黄芩;图2当中,20号峰白头翁皂苷b4来源于白头翁。(b)未鉴定的共有峰3、4、6、7、9、11、14和19峰来源于黄芩,8和10号峰来源于虎杖,16号峰来源于苦参,17号峰来源于板蓝根,18号峰来源于绵马贯众。(4)根据供试品溶液、混合对照品溶液的hplc谱图,已初步建立了七清败毒颗粒指纹图谱,可为其质量控制提供参考。
[0108]
实施例2
[0109]
建立各成分的线性回归方程
[0110]
本实施例当中,hplc的检测参数如下:c-18色谱柱为固定相,以乙腈为流动相a相,0.1%磷酸水溶液为流动相b,检测波长分别为201nm、245nm,柱温为30℃,流速为1ml/min,进样体积为10μl,流动相梯度如表1所示。
[0111]
取实施例1当中的1-6号不同浓度的混合对照品溶液,以上述液相色谱检测参数进行液相色谱分析。采集的液相色谱图谱,根据实施例1当中的方法确定(r,s)-告依春、黄芩苷、黄芩素、汉黄芩素、白头翁皂苷b4、虎杖苷、大黄素谱峰的保留时间,并记录上述各组分的谱峰面积,以每种组分的谱峰面积为纵坐标(y),以及混合对照品中每种组分的浓度为横坐标(x),建立线性回归方程,考察7种组分的含量线性范围。结果如下表2所示:
[0112]
表2 7种组分及其含量的线性指标
[0113][0114]
实施例3
[0115]
不同种类和不同批次的七清败毒颗粒成分及其含量的测定
[0116]
实验室自制七清败毒颗粒样品:取黄芩中药配方颗粒0.4g,大青叶中药配方颗粒0.175g、绵马贯众中药配方颗粒0.087g、虎杖中药配方颗粒0.133g、白头翁中药配方颗粒0.0875g、苦参中药配方颗粒0.175g、板蓝根中药配方颗粒0.266g,加甲醇40ml,加热回流提取30min,滤过,滤液蒸干,残渣中加80%甲醇溶解,并用80%甲醇定容至25ml,作为实验室
自制七清败毒颗粒样品溶液。
[0117]
对5种批次(来源于广东容大生物股份有限公司,批号2006007)和实验室自制的七清败毒颗粒样品的7种成分进行含量测定,其中,不同批次的七清败毒颗粒hplc测试样品溶液的制备方法参考实施例1中供试品溶液的制备方法。其中的液相色谱检测参数如下:以c-18色谱柱为固定相,乙腈为流动相的a相,0.1%磷酸的水溶液为流动相b相,进行梯度洗脱,检测波长分别为201nm、245nm,柱温为30℃,流速为1ml/min,流动相梯度如下表1所示。根据hplc检测结果分析不同批次的七清败毒颗粒成分及含量,结果见表3所示:
[0118]
表3 5种批次七清败毒颗粒和实验室自制七清败毒颗粒的7种成分及其含量
[0119][0120][0121]
一方面,从检测结果的谱图可知,液相色谱的基线稳定,分析时间短,峰型对称,专属性强。另一方面,根据检测结果可知,七清败毒颗粒处方含有的七种药材(黄芩、虎杖、白头翁、苦参、板蓝根、绵马贯众、大青叶),使用本发明的方法有4种药材的7个特征成分可以实现定量分析,分别为黄芩的黄芩苷、黄芩素和汉黄芩素,白头翁的白头翁皂苷b4,虎杖的虎杖苷和大黄素,板蓝根的(r,s)-告依春。苦参(16号峰)和绵马贯众(18号峰)在此方法下也有特征吸收峰可供质量控制参考。
[0122]
苦参的特征成分苦参碱和氧化苦参碱在图谱中并未显示,可能是在此酸性流动相条件下苦参碱和氧化苦参碱成盐,c-18色谱柱对其保留能力降低。大青叶特征成分靛玉红的极性较小,该样品在此处理条件下不能有效富集靛玉红,大青叶对照药材溶液在该色谱条件下无明显吸收峰。
[0123]
由此可见,根据本发明的方法,能够方便快捷地检测同种类且不同批次或者不同种类的七清败毒颗粒的成分及含量,为七清败毒颗粒的质量评估提供可靠的、可量化的标准。
[0124]
最后所应当说明的是,以上实施例仅用以说明本发明的技术方案而非对本发明保护范围的限制,尽管参照较佳实施例对本发明作了详细说明,本领域的普通技术人员应当理解,可以对本发明的技术方案进行修改或者等同替换,而不脱离本发明技术方案的实质和范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。