1.本发明属于催化剂制备技术领域,具体涉及一种含氧化钴载体的钯基催化剂及其制备方法与应用。
背景技术:
2.作为天然气和煤矿瓦斯的主要成分,甲烷具有强烈的温室效应,其效力是co2的21倍。天然气汽车尾气、天然气工业、矿井排风等每天产生大量低浓度(浓度在5%以下)甲烷。5%低于甲烷燃烧的最低浓度界限,使用常规火焰燃烧技术难以将低浓度甲烷完全氧化,如被直接排入大气中会加剧大气温室效应、威胁我们的生存环境。甲烷均相燃烧需要的总活化能很高,在传统的火焰燃烧中,甲烷在》1300℃的高温度下才能发生氧化断裂,同时产生大量的有毒副产物(no和co等)。甲烷可以在贵金属、过渡金属催化剂上以更低的温度进行燃烧转化、且基本不会生成no。因此,可以在较低温度下通过催化燃烧来消除低浓度甲烷。
3.迄今为止开发的甲烷燃烧催化剂主要有负载型贵金属和非贵金属氧化物。近年来,国内外对低浓度甲烷催化燃烧催化剂的研究主要集中于提高催化剂的低温用于低浓度甲烷催化燃烧的高效负载型贵金属纳米催化剂活性、热稳定性、抗毒性和降低催化剂的成本。贵金属氧化物具有较低的起燃温度、较好的稳定性及较好的抗中毒性,因此成为低浓度甲烷催化燃烧催化剂的研究热点。
4.cn106492824a公开了一种甲烷燃烧催化剂,具有核壳结构,其结构为[pd]
0.010
[co3o4]
0.300
@[sio2]
0.690
,核心部分包含四氧化三钴和贵金属,壳层部分包含二氧化硅,用一层耐高温的壳层将贵金属包覆在里面,壳层具有孔道可以让反应分子自由进入核心处与贵金属活性中心发生反应,然后生成的产物分子扩散出壳层,整个过程活性中心的尺寸要大于壳层反应孔道的尺寸,以避免活性组分的溢出,提高了甲烷燃烧催化剂的抗高温性能以及催化燃烧性能,解决了现有技术中甲烷燃烧催化剂存在的高温易烧结的问题。
[0005]
然而,上述方法中的催化效率仍然有待提高。因此,需要提供一种新的催化剂,在高空速以及水蒸气条件下都保持良好的催化性能。为实现甲烷的低温活化和稳定转化,开发一种低负载量、高活性和稳定性的钯基催化剂具有很大的工业应用意义。
技术实现要素:
[0006]
本发明的目的在于提供一种含氧化钴载体的钯基催化剂及其制备方法与应用,通过溶剂热法制备了co3o4载体,并通过浸渍法负载低浓度的贵金属pd,充分利用金属pd与co3o4载体之间的协同作用,提高催化活性以及稳定性;通过调控第二溶剂与第一溶剂的比例以及表面活性剂的用量,实现催化性能的最大化提升;且制备工艺简单,操作方便,绿色高效。
[0007]
为达到此发明目的,本发明采用以下技术方案:
[0008]
本发明的目的之一在于提供一种含氧化钴载体的钯基催化剂的制备方法,所述制备方法包括如下步骤:
[0009]
(1)将表面活性剂与第一溶剂混合后,加入钴源,得到初始溶液;
[0010]
其中,所述表面活性剂包括ctab和/或pvp;
[0011]
(2)将步骤(1)所述初始溶液与第二溶剂混合后进行溶剂热反应,依次经过第一次固液分离和第一焙烧,得到co3o4载体;
[0012]
(3)将步骤(2)所述co3o4载体在钯源溶液中进行浸渍,依次经过第二次固液分离和第二焙烧,得到含氧化钴载体的钯基催化剂。
[0013]
本发明中,通过溶剂热法制备了co3o4载体,并通过浸渍法负载低浓度的贵金属pd,充分利用金属pd与co3o4载体之间的协同作用,提高催化活性以及稳定性;通过调控第二溶剂与第一溶剂的比例以及表面活性剂的用量,实现催化性能的最大化提升;且制备工艺简单,操作方便,绿色高效;
[0014]
值得说明的是,钴源、表面活性剂溶解在第一溶剂中,第二溶剂包覆在外层形成一层碳层保护层,使得焙烧过程中,钴前体缓慢从碳层内释放,减少了焙烧过程导致的团聚,使得焙烧得到的co3o4拥有较多的氧空位,有利于pdo-pd之间的转换,提高了催化性能。
[0015]
作为本发明优选的技术方案,步骤(1)所述表面活性剂为ctab。
[0016]
优选地,步骤(1)所述第一溶剂包括乙醇。
[0017]
优选地,步骤(1)所述钴源包括乙酸钴。
[0018]
作为本发明优选的技术方案,步骤(1)所述钴源中的钴离子与所述表面活性剂的摩尔比为(10-30):1,例如可以是10:1,12:1,15:1,18:1,20:1,23:1,25:1,27:1,30:1等,进一步优选为(17-25):1,例如可以是17:1,18:1,19:1,20:1,21:1,22:1,23:1,24:1,25:1等,但并不仅限于所列举的数值,上述数值范围内其他未列举的数值同样适用。
[0019]
值得说明的是,步骤(1)所述钴源中的钴离子与所述表面活性剂的摩尔比为(10-30):1,若表面活性剂过少,则会不利于钴源分散,进而导致催化性能下降;若表面活性剂过多,则会不利于钴源表面的保护层的形成,进而导致催化性能下降。
[0020]
优选地,步骤(1)所述钴源与所述第一溶剂的质量比为1:(10-15),例如可以是1:10,1:11,1:12,1:13,1:14,1:15等,但并不仅限于所列举的数值,上述数值范围内其他未列举的数值同样适用。
[0021]
作为本发明优选的技术方案,步骤(2)所述第二溶剂包括乙二醇、异丙醇、1,3-丙二醇或甘油中的任意一种或至少两种的组合,所述组合典型但非限制性的实例包括乙二醇和异丙醇的组合,乙二醇和1,3-丙二醇的组合,乙二醇和甘油的组合,异丙醇和1,3-丙二醇的组合,异丙醇和甘油的组合,1,3-丙二醇和甘油的组合;进一步优选为异丙醇。
[0022]
优选地,步骤(2)所述第二溶剂与步骤(1)所述第一溶剂的体积比为1:(2-16),例如可以是1:2,1:3,1:5,1:7,1:8,1:10,1:11,1:12,1:13,1:15,1:16等,但并不仅限于所列举的数值,上述数值范围内其他未列举的数值同样适用。
[0023]
值得说明的是,步骤(2)所述第二溶剂与步骤(1)所述第一溶剂的体积比为1:(2-16),若第二溶剂过多,则会导致钴源的溶解不完全,进而导致催化性能下降;若第二溶剂过少,则不利于保护层的形成,进而导致催化性能下降。
[0024]
优选地,步骤(2)所述溶剂热反应的温度为120-180℃,例如可以是120℃,125℃,130℃,135℃,140℃,145℃,150℃,155℃,160℃,165℃,170℃,175℃,180℃等,但并不仅限于所列举的数值,上述数值范围内其他未列举的数值同样适用。
[0025]
优选地,步骤(2)所述溶剂热反应的时间为4-36h,例如可以是4h,5h,8h,10h,12h,15h,18h,20h,22h,24h,26h,28h,30h,32h,34h,36h等,但并不仅限于所列举的数值,上述数值范围内其他未列举的数值同样适用。
[0026]
作为本发明优选的技术方案,步骤(2)所述第一次固液分离的方式为离心。
[0027]
优选地,步骤(2)所述第一焙烧的温度为600-800℃,例如可以是600℃,620℃,640℃,660℃,680℃,700℃,720℃,740℃,760℃,780℃,800℃等,但并不仅限于所列举的数值,上述数值范围内其他未列举的数值同样适用。
[0028]
优选地,步骤(2)所述第一焙烧的升温速率为2-5℃/min,例如可以是2℃/min,2.5℃/min,3℃/min,3.5℃/min,4℃/min,4.5℃/min,5℃/min等,但并不仅限于所列举的数值,上述数值范围内其他未列举的数值同样适用。
[0029]
优选地,步骤(2)所述第一焙烧的时间为1-12h,例如可以是1h,2h,3h,4h,5h,6h,7h,8h,9h,10h,11h,12h等,但并不仅限于所列举的数值,上述数值范围内其他未列举的数值同样适用。
[0030]
作为本发明优选的技术方案,步骤(3)所述钯源溶液中钯源的含量为0.02-0.03wt%,例如可以是0.02wt%,0.021wt%,0.022wt%,0.023wt%,0.024wt%,0.025wt%,0.026wt%,0.027wt%,0.028wt%,0.029wt%,0.03wt%等,但并不仅限于所列举的数值,上述数值范围内其他未列举的数值同样适用。
[0031]
优选地,以1gco3o4载体为基准,步骤(3)所述钯源溶液的用量为90-120ml,例如可以是90ml,95ml,100ml,105ml,110ml,115ml,120ml等,但并不仅限于所列举的数值,上述数值范围内其他未列举的数值同样适用。
[0032]
优选地,步骤(3)所述浸渍的温度为10-30℃,例如可以是10℃,12℃,14℃,16℃,18℃,20℃,22℃,24℃,26℃,28℃,30℃等,但并不仅限于所列举的数值,上述数值范围内其他未列举的数值同样适用。
[0033]
优选地,步骤(3)所述浸渍的时间为1-2h,例如可以是1h,1.1h,1.2h,1.3h,1.4h,1.5h,1.6h,1.7h,1.8h,1.9h,2h等,但并不仅限于所列举的数值,上述数值范围内其他未列举的数值同样适用。
[0034]
作为本发明优选的技术方案,步骤(3)所述第二次固液分离的方式为旋蒸。
[0035]
优选地,步骤(3)所述第二焙烧的温度为550-650℃,例如可以是550℃,560℃,570℃,580℃,590℃,600℃,610℃,620℃,630℃,640℃,650℃等,但并不仅限于所列举的数值,上述数值范围内其他未列举的数值同样适用。
[0036]
优选地,步骤(3)所述第二焙烧的升温速率为2-5℃/min,例如可以是2℃/min,2.5℃/min,3℃/min,3.5℃/min,4℃/min,4.5℃/min,5℃/min等,但并不仅限于所列举的数值,上述数值范围内其他未列举的数值同样适用。
[0037]
优选地,步骤(3)所述第二焙烧的时间为4-6h,例如可以是4h,4.2h,4.4h,4.6h,4.8h,5h,5.2h,5.4h,5.6h,5.8h,6h等,但并不仅限于所列举的数值,上述数值范围内其他未列举的数值同样适用。
[0038]
作为本发明优选的技术方案,所述制备方法包括如下步骤:
[0039]
(1)将表面活性剂与乙醇混合后,加入乙酸钴,得到初始溶液;
[0040]
其中,表面活性剂包括ctab和/或pvp;乙酸钴中的钴离子与表面活性剂的摩尔比
为(10-30):1;乙酸钴与乙醇的质量比为1:(10-15);
[0041]
(2)将步骤(1)所述初始溶液与第二溶剂混合后在120-180℃进行溶剂热反应4-36h,经过离心后以2-5℃/min的速率升温至600-800℃第一焙烧1-12h,得到co3o4载体;
[0042]
其中,第二溶剂包括乙二醇、异丙醇、1,3-丙二醇或甘油中的任意一种或至少两种的组合;所述第二溶剂与步骤(1)中乙醇的体积比为1:(2-16);
[0043]
(3)将步骤(2)所述co3o4载体在0.02-0.03wt%的硝酸钯溶液中进行浸渍1-2h,以1gco3o4载体为基准,硝酸钯溶液的用量为90-120ml;经过旋蒸后以2-5℃/min的速率升温至550-650℃第二焙烧4-6h,得到含氧化钴载体的钯基催化剂。
[0044]
本发明的目的之二在于提供一种目的之一所述制备方法得到的含氧化钴载体的钯基催化剂,所述钯基催化剂包括co3o4载体和负载在所述co3o4载体上的活性组分pd;所述活性组分pd的含量为0.8-1.2wt%,例如可以是0.8wt%,0.82wt%,0.86wt%,0.9wt%,0.94wt%,0.97wt%,1.0wt%,1.13wt%,1.18wt%,1.2wt%等,但并不仅限于所列举的数值,上述数值范围内其他未列举的数值同样适用。
[0045]
本发明的目的之三在于提供一种目的之二所述含氧化钴载体的钯基催化剂的应用,所述含氧化钴载体的钯基催化剂用于甲烷燃烧反应。
[0046]
值得说明的是,本发明所述含氧化钴载体的钯基催化剂催化甲烷燃烧反应时,原料气包括甲烷、氧气、二氧化碳、水蒸气和氮气,甲烷浓度为800-1200ppm,氧气浓度为3-5%,二氧化碳浓度为5-10%,水蒸气含量为5-10%,其余为氮气。
[0047]
本发明所述的数值范围不仅包括上述例举的点值,还包括没有例举出的上述数值范围之间的任意的点值,限于篇幅及出于简明的考虑,本发明不再穷尽列举所述范围包括的具体点值。
[0048]
相对于现有技术,本发明具有以下有益效果:
[0049]
(1)本发明所述含氧化钴载体的钯基催化剂,以还原性co3o4为载体,负载低浓度的贵金属pd,充分利用金属pd与co3o4载体之间的协同作用,提高催化活性以及稳定性,且催化剂可以回收循环使用,降低了成本;
[0050]
(2)本发明所述含氧化钴载体的钯基催化剂的制备方法中,通过调控第二溶剂与第一溶剂的比例以及表面活性剂的用量,实现催化性能的最大化提升;且工艺简单,操作方便,绿色高效。
附图说明
[0051]
图1为实施例1-3和对比例1所得催化剂在不同温度下催化甲烷燃烧的甲烷转化率曲线;
[0052]
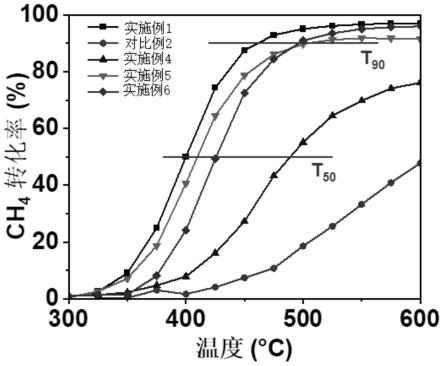
图2为实施例1、4-6和对比例2所得催化剂在不同温度下催化甲烷燃烧的甲烷转化率曲线;
[0053]
图3为实施例1所得催化剂的稳定性曲线。
具体实施方式
[0054]
下面通过具体实施方式来进一步说明本发明的技术方案。本领域技术人员应该明了,所述实施例仅仅是帮助理解本发明,不应视为对本发明的具体限制。
[0055]
实施例1
[0056]
本实施例提供了一种含氧化钴载体的钯基催化剂及其制备方法,所述钯基催化剂包括co3o4载体和负载在所述co3o4载体上的活性组分pd;所述活性组分pd的含量为1wt%;
[0057]
所述制备方法包括如下步骤:
[0058]
(1)将ctab与乙醇混合后,加入乙酸钴,得到初始溶液;
[0059]
其中,乙酸钴中的钴离子与ctab的摩尔比为20:1;乙酸钴与乙醇的质量比为1:12.5;
[0060]
(2)将步骤(1)所述初始溶液与异丙醇混合后在160℃进行溶剂热反应12h,经过离心后以2℃/min的速率升温至700℃第一焙烧5h,得到co3o4载体;
[0061]
其中,异丙醇与步骤(1)中乙醇的体积比为1:4;
[0062]
(3)将步骤(2)所述co3o4载体在0.025wt%的硝酸钯溶液中进行浸渍1h,以1g co3o4载体为基准,硝酸钯溶液的用量为90ml;经过旋蒸后以2℃/min的速率升温至600℃第二焙烧5h,得到含氧化钴载体的钯基催化剂。
[0063]
实施例2
[0064]
本实施例提供了一种含氧化钴载体的钯基催化剂及其制备方法,所述钯基催化剂包括co3o4载体和负载在所述co3o4载体上的活性组分pd;所述活性组分pd的含量为1wt%;
[0065]
所述制备方法参照实施例1所述的制备方法,区别仅在于:步骤(1)中乙酸钴中的钴离子与ctab的摩尔比为10:1。
[0066]
实施例3
[0067]
本实施例提供了一种含氧化钴载体的钯基催化剂及其制备方法,所述钯基催化剂包括co3o4载体和负载在所述co3o4载体上的活性组分pd;所述活性组分pd的含量为1wt%;
[0068]
所述制备方法参照实施例1所述的制备方法,区别仅在于:步骤(1)中乙酸钴中的钴离子与ctab的摩尔比为30:1。
[0069]
实施例4
[0070]
本实施例提供了一种含氧化钴载体的钯基催化剂及其制备方法,所述钯基催化剂包括co3o4载体和负载在所述co3o4载体上的活性组分pd;所述活性组分pd的含量为1wt%;
[0071]
所述制备方法参照实施例1所述的制备方法,区别仅在于:步骤(2)中异丙醇与步骤(1)中乙醇的体积比为1:2。
[0072]
实施例5
[0073]
本实施例提供了一种含氧化钴载体的钯基催化剂及其制备方法,所述钯基催化剂包括co3o4载体和负载在所述co3o4载体上的活性组分pd;所述活性组分pd的含量为1wt%;
[0074]
所述制备方法参照实施例1所述的制备方法,区别仅在于:步骤(2)中异丙醇与步骤(1)中乙醇的体积比为1:8。
[0075]
实施例6
[0076]
本实施例提供了一种含氧化钴载体的钯基催化剂及其制备方法,所述钯基催化剂包括co3o4载体和负载在所述co3o4载体上的活性组分pd;所述活性组分pd的含量为1wt%;
[0077]
所述制备方法参照实施例1所述的制备方法,区别仅在于:步骤(2)中异丙醇与步骤(1)中乙醇的体积比为1:16。
[0078]
实施例7
[0079]
本实施例提供了一种含氧化钴载体的钯基催化剂及其制备方法,所述钯基催化剂包括co3o4载体和负载在所述co3o4载体上的活性组分pd;所述活性组分pd的含量为1.2wt%;
[0080]
所述制备方法包括如下步骤:
[0081]
(1)将ctab与乙醇混合后,加入乙酸钴,得到初始溶液;
[0082]
其中,乙酸钴中的钴离子与ctab的摩尔比为20:1;乙酸钴与乙醇的质量比为1:10;
[0083]
(2)将步骤(1)所述初始溶液与乙二醇混合后在120℃进行溶剂热反应24h,经过离心后以3℃/min的速率升温至600℃第一焙烧12h,得到co3o4载体;
[0084]
其中,所述乙二醇与步骤(1)中乙醇的体积比为1:4;
[0085]
(3)将步骤(2)所述co3o4载体在0.03wt%的硝酸钯溶液中进行浸渍2h,以1gco3o4载体为基准,硝酸钯溶液的用量为120ml;经过旋蒸后以3℃/min的速率升温至550℃第二焙烧6h,得到含氧化钴载体的钯基催化剂。
[0086]
实施例8
[0087]
本实施例提供了一种含氧化钴载体的钯基催化剂及其制备方法,所述钯基催化剂包括co3o4载体和负载在所述co3o4载体上的活性组分pd;所述活性组分pd的含量为0.8wt%;
[0088]
所述制备方法包括如下步骤:
[0089]
(1)将ctab与乙醇混合后,加入乙酸钴,得到初始溶液;
[0090]
其中,乙酸钴中的钴离子与ctab的摩尔比为20:1;乙酸钴与乙醇的质量比为1:15;
[0091]
(2)将步骤(1)所述初始溶液与异丙醇混合后在180℃进行溶剂热反应4h,经过离心后以5℃/min的速率升温至800℃第一焙烧1h,得到co3o4载体;
[0092]
其中,异丙醇与步骤(1)中乙醇的体积比为1:4;
[0093]
(3)将步骤(2)所述co3o4载体在0.02wt%的硝酸钯溶液中进行浸渍1.5h,以1gco3o4载体为基准,硝酸钯溶液的用量为100ml;经过旋蒸后以5℃/min的速率升温至650℃第二焙烧4h,得到含氧化钴载体的钯基催化剂。
[0094]
对比例1
[0095]
本对比例提供了一种含氧化钴载体的钯基催化剂及其制备方法,所述钯基催化剂包括co3o4载体和负载在所述co3o4载体上的活性组分pd;所述活性组分pd的含量为1wt%;
[0096]
所述制备方法参照实施例1所述的制备方法,区别仅在于:步骤(1)中未加入ctab。
[0097]
对比例2
[0098]
本对比例提供了一种含氧化钴载体的钯基催化剂及其制备方法,所述钯基催化剂包括co3o4载体和负载在所述co3o4载体上的活性组分pd;所述活性组分pd的含量为1wt%;
[0099]
所述制备方法参照实施例1所述的制备方法,区别仅在于:步骤(2)中未加入异丙醇。
[0100]
(一)将上述实施例与对比例所得含氧化钴载体的钯基催化剂的催化性能进行测试,方法如下:
[0101]
将0.24g钯基催化剂装填在连续流动微型固定床上,以ghsv=300,000ml/h
·
g的质量空速通入混合气,混合气的组成为:1000ppm的ch4,3.5vol.%的o2,6vol.%的co2,10%的h2o,n2平衡;通过红外烟气分析仪测定尾气中甲烷浓度随温度的变化;
[0102]
将上述实施例与对比例催化甲烷燃烧时转化率分别达到50%和90%时的温度t50和t90,以及在500℃的甲烷转化率列于表1。
[0103]
将实施例1-3和对比例1所得催化剂催化甲烷燃烧时不同温度下的甲烷转化率曲线列于图1,由图1可以看出,当co
2
和ctab的摩尔比为20:1时,催化剂的催化性能最佳。
[0104]
将实施例1、4-6和对比例2所得催化剂催化甲烷燃烧时不同温度下的甲烷转化率曲线列于图2,由图2可以看出,当异丙醇和乙醇的体积比为4:1时,催化剂的催化性能最佳。
[0105]
(二)将实施例1所得含氧化钴载体的钯基催化剂的稳定性进行测试,测试方法如下:
[0106]
将0.24g钯基催化剂装填在连续流动微型固定床上,控制温度在475℃,保温12h,以ghsv=300,000ml/h
·
g的质量空速通入混合气,混合气的组成为:1000ppm的ch4,3.5vol.%的o2,6vol.%的co2,10%的h2o,n2平衡;通过气相色谱测定12h内尾气中甲烷的浓度变化;
[0107]
将实施例1稳定性测试的结果列于图3,由图3可以看出,在12h内,催化剂的催化性能保持稳定,尾气中甲烷的浓度几乎不发生变化。
[0108]
表1
[0109][0110][0111]
由表1可以得出以下几点:
[0112]
(1)由实施例1、7-8可以看出,本发明通过充分利用金属pd与co3o4载体之间的协同作用,提高催化活性以及稳定性,以还原性co3o4为载体,有助于pdo-pd之间的转换,提升催
化性能;
[0113]
(2)由实施例1-3、对比例1的比较,可以看出,实施例1钴离子与ctab的摩尔比为20:1,其低温催化性能优异,t50为400℃,t90为460℃,500℃的甲烷转化率达到95%,由于ctab可改善载体的表面形貌和晶面暴露,进而催化性能优异;相较于实施例1,实施例2钴离子与ctab的摩尔比为10:1,其催化性能下降,t50和t90均高于实施例1,500℃的甲烷转化率低于实施例1;相较于实施例1,实施例3钴离子与ctab的摩尔比为30:1,其催化性能下降,t50和t90均高于实施例1,500℃的甲烷转化率低于实施例1;相较于实施例1,对比例1未加入ctab,导致其催化性能下降;
[0114]
(3)由实施例1、4-6、对比例2的比较,可以看出,实施例1的步骤(2)中异丙醇与步骤(1)中乙醇的体积比为1:4,其低温催化性能优异,异丙醇的加入,可减少co3o4前驱体在焙烧过程中的团聚;相较于实施例1,实施例4的步骤(2)中异丙醇与步骤(1)中乙醇的体积比为1:2,其催化性能下降,t50和t90均高于实施例1,500℃的甲烷转化率低于实施例1;相较于实施例1,实施例5的步骤(2)中异丙醇与步骤(1)中乙醇的体积比为1:8,其催化性能下降,t50和t90均高于实施例1,500℃的甲烷转化率低于实施例1;相较于实施例1,实施例6的步骤(2)中异丙醇与步骤(1)中乙醇的体积比为1:16,其催化性能下降,t50和t90均高于实施例1,500℃的甲烷转化率低于实施例1;相较于实施例1,对比例2未加入异丙醇,导致其催化性能大幅下降。
[0115]
申请人声明,以上所述仅为本发明的具体实施方式,但本发明的保护范围并不局限于此,所属技术领域的技术人员应该明了,任何属于本技术领域的技术人员在本发明揭露的技术范围内,可轻易想到的变化或替换,均落在本发明的保护范围和公开范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。