1.本发明涉及一种含低氧型纳米硅粒子的浆料、负极活性物质、负极及锂离子二次电池。
背景技术:
2.近年来,随着智能手机等便携式电子设备的普及,小型、高容量二次电池的需求提高。其中,锂离子二次电池(有时记载为lib)正在推进向电动汽车(ev)的急速展开,产业上的利用范围正在扩大。作为锂离子二次电池的负极材料,广泛使用碳类的石墨活性物质(天然、人工),但石墨的理论容量密度低(372mah/g),随着锂离子二次电池构成技术的进化,电池容量提高接近极限。
3.因此,为了锂离子二次电池的高容量、高能量密度化,正在研究由使用了与锂离子合金化的硅和它们的氧化物的硅系材料构成的活性物质。其中,碳氧化硅(以下,有时表述为sioc)是由陶瓷骨架和自由碳构成的材料,该陶瓷骨架由si、o和c构成,与其他种类的高容量活性物质相比具有优异的充放电循环特性,从而受到关注。但是,由于结构上的限制,具有充放电容量和首次库仑效率均低的弱点,因此sioc尚未达到实用。为了解决该课题,研究了将sioc与硅、硅合金或氧化硅复合化的方法。例如,在专利文献1中,提出了一种锂离子二次电池用负极材料,其由复合粒子构成,该复合粒子相对于sioc含有5~30体积%的活性物质粒子,该活性物质粒子由硅粒子或由碳被覆的硅粒子构成。另外,还公开了将硅、硅合金或氧化硅的微粒与作为无机粘合剂的sioc复合化,并且导入了球状或鳞片状石墨的硅系无机氧化物复合体粒子(专利文献2)。
4.然而,已知专利文献1、2中导入的硅、硅合金在充放电时会由于体积膨胀、收缩的反复而微粉化,发生电极材料从集电体剥离、崩解、电子传导性的恶化等,从而导致充放电循环特性的降低。为了解决这些课题,通常通过将硅粒子粉碎而纳米尺寸化,从而对由反复充放电引起的体积膨胀、收缩所引起的应变进行应力缓和,由此尝试充放电循环特性的改善。然而,存在如下问题:同时引起由纳米化引起的表面积增加和与此相伴的由硅粒子的表面氧化引起的容量降低、库仑效率降低这样的电池特性降低。另外,若将硅粒子粉碎而纳米尺寸化,则在含有该硅粒子的浆料中存在引起粘度增加的课题。
5.现有技术文献
6.专利文献
7.专利文献1:日本特开2016-139579号公报
8.专利文献2:日本特开2005-310759号公报
技术实现要素:
9.发明所要解决的课题
10.鉴于上述实际情况,本发明所要解决的课题在于提供一种含低氧型纳米硅粒子的
浆料,其能够抑制与硅粒子的纳米尺寸化相伴随的增稠,能够用于制造具有优异的充放电特性(充放电容量、首次库仑效率及充放电循环特性)的锂离子二次电池。
11.用于解决课题的手段
12.本发明人等为了解决上述课题,对于在使用硅系无机化合物(例如sioc/c基体)作为负极活性物质的锂离子二次电池中,能够最大限度地提高充放电特性的含纳米硅粒子的浆料进行了反复研究,结果发现,通过将纳米硅粒子控制为低氧化状态而发挥优异的充放电特性,从而完成了本发明。
13.即,本发明如下所示。
14.项1.一种含低氧型纳米硅粒子的浆料,其含有低氧型纳米硅粒子、非水溶剂和添加剂,
15.前述低氧型纳米硅粒子在
29
si-nmr中处于-100~-110ppm的范围的峰面积(ii)相对于处于-75~-85ppm的范围的峰面积(i)之比[(ii)/(i)比]为1.0以下。
[0016]
项2.如项1所述的含低氧型纳米硅粒子的浆料,其中,前述低氧型纳米硅粒子的体积平均粒径(d50)为10~200nm。
[0017]
项3.根据项1或2所述的含低氧型硅的浆料,其中,作为前述添加剂,包含阳离子性表面活性剂和/或阴离子性表面活性剂。
[0018]
项4.如项3所述的含低氧型硅低氧型硅的浆料,其中,前述添加剂中阳离子性表面活性剂的胺值为1~100mgkoh/g。
[0019]
项5.如项3所述的含低氧型硅低氧型硅的浆料,其中,前述添加剂中阴离子性表面活性剂的酸值为1~200mgkoh/g。
[0020]
项6.如项1~5中任一项所述的含低氧型纳米硅粒子的浆料,其中,前述低氧型纳米硅粒子具有片状。
[0021]
项7.如项1~6中任一项所述的含低氧型纳米硅粒子的浆料,其中,粘度为10mpa
·
s以下。
[0022]
项8.如项1~7中任一项所述的含低氧型纳米硅粒子的浆料,其中,110℃时的不挥发成分为5~40重量%。
[0023]
项9.如项1~8中任一项所述的含低氧型纳米硅粒子的浆料,其中,前述非水溶剂为酮系溶剂。
[0024]
项10.如项1~9中任一项所述的含低氧型纳米硅粒子的浆料,其中,相对于前述低氧型纳米硅粒子100质量份,使用5~60质量份的前述添加剂。
[0025]
项11.一种锂离子二次电池用负极活性物质,其中,原料的一部分包含项1~10中任一项所述的含低氧型纳米硅粒子的浆料。
[0026]
项12.一种二次电池,其在负极中包含项11所述的锂离子二次电池活性物质。
[0027]
项13.项1~10中任一项所述的含低氧型纳米硅粒子的浆料的制造方法,通过使用湿式珠磨机进行分散处理而得到低氧型纳米硅粒子。
[0028]
项14.如项13所述的含低氧型纳米硅粒子的浆料的制造方法,其中,前述分散处理在非活性气体气氛中进行。
[0029]
发明效果
[0030]
本发明的含低氧型纳米硅粒子的浆料中的低氧型纳米硅粒子作为负极活性物质,
均匀地分散于硅系无机化合物(例如sioc/c基体)中从而抑制膨胀率,因此充放电循环特性优异,且抑制了硅表面的氧化,由此充放电容量与初始库仑效率高。另外,本发明的含低氧型纳米硅粒子的浆料能够兼顾硅的纳米尺寸化和低粘度。
附图说明
[0031]
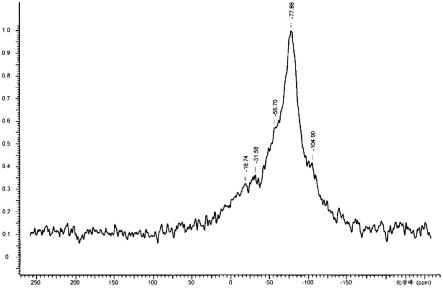
[图1]是实施例1的低氧型纳米硅粒子的
29
si-nmr光谱的图表。
[0032]
[图2]是实施例1的低氧型纳米硅粒子的透射型电子显微镜(tem)图像。
[0033]
[图3]是实施例2的低氧型纳米硅粒子的
29
si-nmr光谱的图表。
[0034]
[图4]是实施例3的低氧型纳米硅粒子的
29
si-nmr光谱的图表。
[0035]
[图5]是实施例3的低氧型纳米硅粒子的透射型电子显微镜(tem)图像。
[0036]
[图6]是实施例4的低氧型纳米硅粒子的
29
si-nmr光谱的图表。
[0037]
[图7]是比较例1的低氧型纳米硅粒子的透射型电子显微镜(tem)图像。
具体实施方式
[0038]
《含低氧型纳米硅粒子的浆料》
[0039]
本发明的含低氧型纳米硅粒子的浆料含有低氧型纳米硅粒子、非水溶剂和添加剂。以下,对各成分进行说明。需要说明的是,含低氧型纳米硅粒子的浆料的制造方法如后述的锂离子二次电池负极活性物质的制造方法中的工序1所示。
[0040]
(低氧型纳米硅粒子)
[0041]
就低氧型纳米硅粒子而言,在
29
si-nmr中,处于-100~-110ppm的范围的峰面积(ii)相对于处于-75~-85ppm的范围的峰面积(i)之比[(ii)/(i)比]为1.0以下。上述峰面积(i)源自si(0价),另外峰面积(ii)源自sio2、sio4等si(0价以外)。作为该峰面积比的(ii)/(i)比为1.0以下是指在纳米硅粒子中si(0价)的比例比si(0价以外)多,这表示在纳米硅粒子中未进行氧化(为低氧化状态)。如此,通过使纳米硅粒子为低氧化状态,可有效地抑制制成电池时的容量降低、库伦效率降低等电池特性的降低。另外,作为峰面积比的(ii)/(i)比优选为0.01~0.8,更优选为0.01~0.5。
[0042]
低氧型纳米硅粒子的
29
si-nmr光谱是使用固体nmr装置可容易地得到的,本说明书的固体nmr测定是使用日本电子株式会社jeol制装置(jnm-eca600)实施的。
[0043]
作为上述峰面积比的(ii)/(i)比可以通过以下的方法算出。关于上述峰面积比,利用固体nmr分析装置进行单脉冲测定,对得到的固体nmr光谱数据进行傅里叶变换,使用高斯(gauss) 洛伦兹(lorentz)函数对其进行波形分离。接着,基于通过波形分离所得到的峰面积,求出处于-100~-110ppm的范围的峰面积(ii)相对于处于-75~-85ppm的范围的峰面积(i)的比[(ii)/(i)比]。
[0044]
低氧型纳米硅粒子的体积平均粒径(d50)优选为10~200nm,更优选为10~100nm,进一步优选为20~80nm。低氧型纳米硅粒子的体积平均粒径(d50)可以使用激光粒度分析仪等通过动态光散射法来测定。超过200nm的大尺寸的硅粒子成为大块,在充放电时容易引起微粉化现象,因此可设想活性物质的充放电性能降低的倾向。另一方面,小于10nm的小尺寸的硅粒子过细,因此硅粒子彼此容易凝聚。因此,难以使小粒子硅均匀地分散于活性物质中,另外,还存在微小粒子的表面活性能高、在活性物质的高温烧成中副产物等在小粒子硅
的表面上增多的倾向,这会导致充放电性能的大幅降低。
[0045]
低氧型纳米硅粒子优选具有片状,另外优选长轴方向的长度为50~300nm且厚度为1~60nm。在本发明中,片状特别是指厚度/长度(所谓的纵横比)为0.5以下。纵横比超过0.5的大尺寸的硅粒子成为大块,在充放电时容易引起微粉化现象,因此可设想活性物质的充放电性能降低的倾向。
[0046]
具有片状的纳米硅粒子的形态可以利用动态光散射法测定平均粒径,但通过使用透射型电子显微镜(tem)、场发射扫描电子显微镜(fe-sem)的解析手段,可以更容易且精密地鉴定上述那样的厚度/长度等样品形态(尺寸、形状等)。需要说明的是,在内包有片状硅纳米粒子的负极活性物质粉末的情况下,可以用聚焦离子束(fib)切断样品而对截面进行fe-sem观察,或者可以对样品进行切片加工并通过tem观察来鉴定硅粒子的状态。
[0047]
本发明中规定的低氧型纳米硅粒子的尺寸范围是以tem图像中显现的视野内的样品的主要部分50个粒子为基础的计算结果。由于也存在观察视野的极限,因此本发明中的低氧型纳米硅粒子也可以具有偏离上述范围的尺寸。
[0048]
低氧型纳米硅粒子可以在表面上存在氧化硅的薄膜,也可以被氧化硅以外的金属氧化物覆盖。对该金属氧化物的种类没有特别限定,可以举出氧化钛、氧化锰、氧化铝、氧化锌等。关于氧化物薄膜的厚度,只要在充放电时不阻止锂离子的传导和电子的迁移就没有特别限定,优选为10nm以下。
[0049]
(非水溶剂)
[0050]
作为非水溶剂,为了抑制由金属硅与水的反应引起的氧化,优选烃系、酮系、酯系、醚系的溶剂。特别是从能够抑制硅表面氧化的观点出发,优选mek(甲基醚酮)等酮系溶剂。相对于低氧型纳米硅粒子100质量份,非水溶剂的比例例如为100~1000质量份,优选为200~800质量份。本发明的含低氧型纳米硅粒子的浆料优选以含低氧型纳米硅粒子的浆料的粘度及110℃时的不挥发成分成为后述的范围的比例含有非水溶剂。
[0051]
(添加剂)
[0052]
作为添加剂,优选阳离子性表面活性剂、阴离子性表面活性剂、两性表面活性剂。作为阳离子性表面活性剂,可举出具有酰胺基、伯胺、仲胺、叔胺、亚胺、烯胺等作为极性基团的表面活性剂。作为阴离子性表面活性剂,可以举出具有羧基、磺酸基、硫酸酯基、磷酸酯基等作为极性基团的表面活性剂。其中,作为添加剂,优选包含阳离子性表面活性剂和/或阴离子性表面活性剂。
[0053]
阳离子性表面活性剂中的胺值例如为1~100mgkoh/g,优选为5~80mgkoh/g,更优选为10~48mgkoh/g,特别优选为35~48mgkoh/g。如果胺值为上述范围,则通过粉碎而纳米化的硅粒子彼此的再凝聚得到抑制,因此能够减小浆料的粘度,结果电池的循环特性、充放电容量、初始库仑效率优异。
[0054]
阴离子性表面活性剂的酸值例如为1~200mgkoh/g,优选为10~180mgkoh/g,更优选为50~150mgkoh/g。若酸值为上述范围,则硅粒子对分散介质的润湿性提高,因此可减小浆料的粘度,结果电池的循环特性、充放电容量、初始库仑效率优异。
[0055]
添加剂可以是具有上述胺值和酸值的两性表面活性剂,也可以并用具有上述胺值和酸值的阳离子性和阴离子性的表面活性剂。并用时的含有比率以阳离子性/阴离子性的质量比计特别优选为5/35~35/5。如果以该比例并用表面活性剂,则硅粒子对分散介质的
润湿性提高,纳米化进行,并且通过粉碎而纳米化的硅粒子彼此的再凝聚得到抑制,因此能够减小浆料的粘度,结果电池的循环特性、充放电容量、初始库仑效率优异。
[0056]
相对于低氧型纳米硅粒子100质量份,添加剂的比例例如为5~60质量份,优选为10~40质量份,更优选为20~40质量份。若添加量小于5质量份,则发生硅粒子的凝聚而纳米化不进行,因此无法减小浆料的粘度,结果成为电池的循环特性、充放电容量、初始库仑效率差的结果。另外,即使添加量超过60质量份,上述那样的电池性能也没有大的差异,但粉碎花费时间等在制造时可能产生不良情况。
[0057]
本发明的含低氧型纳米硅粒子的浆料中,除了上述成分以外,作为其他成分,也可以含有硅烷偶联剂、消泡剂、流平剂、粘度调节剂等。
[0058]
本发明的含低氧型纳米硅粒子的浆料的粘度优选为10mpa
·
s以下,更优选为0.5~5mpa
·
s,进一步优选为1.0~4mpa
·
s。本发明的含低氧型纳米硅粒子的浆料通过含有上述添加剂而难以增粘,通过在上述添加剂的存在下将硅粒子粉碎,能够微细化至纳米尺寸。通过将硅粒子微细化至纳米尺寸,硅系无机化合物(例如sioc/c基体)中含有该硅粒子的负极活性物质能够抑制膨胀率,制成电池时的循环特性、充放电容量、初始库仑效率优异。
[0059]
本发明的含低氧型纳米硅粒子的浆料在110℃时的不挥发成分优选为5~40重量%,更优选为8~30重量%,进一步优选为10~25重量%。本发明的含低氧型纳米硅粒子的浆料能够通过添加剂来抑制粘度的上升,因此与以往的浆料相比,能够提高不挥发成分的比例,能够提高制成电池时的循环特性、充放电容量、初始库仑效率。
[0060]
《锂离子二次电池负极活性物质》
[0061]
本发明的锂离子二次电池负极活性物质在原料的一部分中包含上述本发明的含低氧型纳米硅粒子的浆料。另外,本发明的锂离子二次电池负极活性物质也可以包含在
29
si-nmr中处于-100~~110ppm的范围的峰面积(ii)相对于处于-75~~85ppm的范围的峰面积(i)之比[(ii)/(i)比]为1.0以下的低氧型纳米硅粒子。低氧型纳米硅粒子如上所述。
[0062]
本发明的锂离子二次电池负极活性物质优选在包含低氧型纳米硅粒子的同时还包含硅系无机化合物。上述低氧型纳米硅粒子可以添加到公知惯用的负极活性物质中使用。本发明中的锂离子二次电池负极活性物质优选包含上述低氧型纳米硅粒子和其他硅系无机化合物,该低氧型纳米硅粒子具有内包于其他硅系无机化合物中的结构。作为内包硅纳米粒子的其他硅系无机化合物,例如可举出sic(碳化硅)、sioc(氧碳化硅)等。
[0063]
硅系无机化合物中,与氧碳化硅中的si-o-c骨架结构一起存在自由碳。自由碳的导电性优异,同时,如果所含的碳以特定的化学结合状态存在,则碳的结晶质/非晶质碳结构的平衡变好,在硅系无机化合物中,si-o-c骨架结构与自由碳能够三维地形成缠绕结构,因此认为在电池负极中使用时,在充放电时能够灵活地追随低氧型纳米硅[硅(0价)]粒子的体积变化。该自由碳状态可以通过
13
c-nmr测定来评价,使用sp2与sp3的当量比来表示。需要说明的是,在
13
c-nmr测定中,si-o-c骨架结构中的碳由于不存在c-c键而未被检测出,因此能够仅检测自由碳中的碳信息。
[0064]
在本发明的锂离子二次电池负极活性物质中使用的硅系无机化合物中,如上所述,不仅碳的化学结合状态,自由碳的存在量也对充放电特性产生大的影响。如果碳的量不足,则有时导电性差,充放电特性变差。另一方面,如果碳的量过多,则自由碳自身的理论容量低,因此活性物质整体的充放电容量降低。
[0065]
硅系无机化合物中,存在于si-o-c的骨架以外的自由碳容易在大气中热分解,可根据热重损失值算出自由碳的存在量。热分解重量损失通过使用热重差热分析装置thermogravimeter-differential thermal analyzer(tg-dta)而容易地鉴定。另一方面,si-o-c骨架中的“c”具有非常强的化学键,因此热稳定性高,被氧化分解是非常困难的。
[0066]
本发明中的自由碳具有与硬碳类似的结构,随着在大气中在约600℃~900℃的温度范围内被热分解,发生急剧的重量减少。tg-dta测定的最高温度没有特别限定,为了使热分解反应彻底,优选在大气中、不超过1000℃的条件下进行tg-dta测定。根据前述理由,所得到的重量损失的值表示游离碳的存在量。作为本发明中的碳量,在5~60质量%的范围内,更优选为8~50质量%。
[0067]
[锂离子二次电池负极活性物质的制造方法]
[0068]
本发明的锂离子二次电池负极活性物质没有特别限定,例如可以通过以下的工序1~4来制造。
[0069]
工序1:得到本发明的含低氧型纳米硅粒子的浆料的工序
[0070]
工序2:将工序1中得到的含低氧型纳米硅粒子的浆料、聚硅氧烷化合物和碳源树脂混合、分散后,进行干燥,由此得到混合体(前体)的工序
[0071]
工序3:通过将工序2中得到的混合体(前体)在非活性气氛中烧成而得到烧成物的工序
[0072]
工序4:通过将工序3中得到的烧成物粉碎而得到负极活性物质的工序
[0073]
(工序1)
[0074]
对于得到含低氧型纳米硅粒子的浆料的工序,没有特别限定,可以是将市售的金属硅微粒和上述添加剂添加到上述非水溶剂中并通过粉碎加工进行分散处理的方法,也可以是将粉碎加工后的市售的金属硅微粒和上述添加剂添加到上述非水溶剂中并进行分散的方法。关于利用粉碎加工的分散处理,可以是干式粉碎,但湿式粉碎法由于具有在加工时有效地阻止硅粒子的氧化反应的效果,因此优选。对于粉碎装置没有特别限定,可以使用喷射磨、球磨机、珠磨机等。作为粉碎加工,从有效地获得低氧型纳米硅粒子的观点出发,优选使用湿式珠磨机进行分散处理。
[0075]
另外,为了抑制硅粒子的氧化,上述分散处理优选在氮、氩等非活性气体气氛中进行。
[0076]
作为成为低氧型纳米硅粒子的原料的金属硅微粒,优选硅纯度为97%以上,更优选为99.0%以上。添加剂和非水溶剂可以使用上述添加剂和非水溶剂。添加剂及非水溶剂的使用量也如上所述。含低氧型纳米硅粒子的浆料的粘度及110℃时的不挥发成分优选调整为上述范围。得到的低氧型纳米硅粒子的平均粒径优选为10~200nm,更优选为10~100nm,进一步优选为20~80nm。
[0077]
在上述粉碎加工中,为了进一步提高硅粒子的分散性,也可以使用硅烷偶联剂。硅烷偶联剂是在分子内同时具有与有机材料反应结合的官能团和与硅的氧化膜反应结合的官能团的有机硅化合物,通常其结构如下所示:y-r-si-(x)3。在此,y是与有机材料反应结合的官能团,作为其代表例,可举出乙烯基、环氧基、氨基等。x是与硅的氧化膜反应的官能团,通过水或湿气受到水解而生成硅烷醇,该硅烷醇与硅上的氧化膜反应结合。作为x的代表例,可举出烷氧基、乙酰氧基、氯原子等。
[0078]
(工序2)
[0079]
工序2中,将工序1中得到的含低氧型纳米硅粒子的浆料、聚硅氧烷化合物和碳源树脂混合、分散后,进行干燥,由此得到混合体(前体)。
[0080]
作为上述聚硅氧烷化合物,只要是包含至少1个聚碳硅烷、聚硅氮烷、聚硅烷和聚硅氧烷结构的树脂就没有特别限定。可以是这些单独的树脂,也可以是具有其作为链段、与其他聚合物链段化学结合的复合型树脂。作为复合化的形态,有接枝、嵌段、无规、交替等的共聚物。例如,有具有在聚合物链段的侧链上与聚硅氧烷链段化学结合的接枝结构的复合树脂,可列举出在聚合物链段的末端化学结合有聚硅氧烷链段的具有嵌段结构的复合树脂等。
[0081]
上述聚硅氧烷链段优选具有下述通式(s-1)和/或下述通式(s-2)所示的结构单元。
[0082]
[化1]
[0083][0084]
[化2]
[0085][0086]
(上述通式(s-1)及(s-2)中,r1表示芳香族烃取代基或烷基,r2及r3分别表示烷基、环烷基、芳基或芳烷基。)
[0087]
作为上述烷基,例如可列举出甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基、叔丁基、戊基、异戊基、新戊基、叔戊基、1-甲基丁基、2-甲基丁基、1,2-二甲基丙基、1-乙基丙基、己基、异己基、1-甲基戊基、2-甲基戊基、3-甲基戊基、1,1-二甲基丁基、1,2-二甲基丁基、2,2-二甲基丁基、1-乙基丁基、1,1,2-三甲基丙基、1,2,2-三甲基丙基、1-乙基-2-甲基丙基、1-乙基-1-甲基丙基等。作为上述环烷基,例如可列举出环丙基、环丁基、环戊基、环己基等。
[0088]
作为上述芳基,例如可列举出苯基、萘基、2-甲基苯基、3-甲基苯基、4-甲基苯基、4-乙烯基苯基、3-异丙基苯基等。
[0089]
作为上述芳烷基,例如可列举出苄基、二苯基甲基、萘基甲基等。
[0090]
作为上述聚硅氧烷化合物所具有的聚硅氧烷链段以外的聚合物链段,例如可列举出丙烯酸系聚合物、氟烯烃聚合物、乙烯基酯聚合物、芳香族系乙烯基聚合物、聚烯烃聚合物等乙烯基聚合物链段、聚氨酯聚合物链段、聚酯聚合物链段、聚醚聚合物链段等聚合物链段等。其中,优选乙烯基聚合物链段。
[0091]
上述聚硅氧烷化合物可以是聚硅氧烷链段与聚合物链段以下述结构式(s-3)所示的结构结合而成的复合树脂,也可以具有三维网眼状的聚硅氧烷结构。
[0092]
[化3]
[0093][0094]
(式中,碳原子为构成聚合物链段的碳原子,2个硅原子为构成聚硅氧烷链段的硅原子。)
[0095]
上述聚硅氧烷化合物所具有的聚硅氧烷链段可以在该聚硅氧烷链段中具有聚合性双键等能够通过加热而反应的官能团。通过在热分解前对聚硅氧烷化合物进行加热处理,从而进行交联反应,形成固体状,由此可以容易地进行热分解处理。
[0096]
作为上述聚合性双键,例如可列举出乙烯基、(甲基)丙烯酰基等。聚合性双键优选在聚硅氧烷链段中存在2个以上,更优选存在3~200个,进一步优选存在3~50个。另外,通过使用存在2个以上聚合性双键的复合树脂作为聚硅氧烷化合物,能够容易地进行交联反应。
[0097]
上述聚硅氧烷链段可以具有硅烷醇基和/或水解性甲硅烷基。作为水解性甲硅烷基中的水解性基团,例如可列举出卤素原子、烷氧基、取代烷氧基、酰氧基、苯氧基、巯基、氨基、酰胺基、氨基氧基、亚氨基氧基、烯氧基等,通过这些基团被水解,水解性甲硅烷基成为硅烷醇基。与前述热固化反应并行地,在硅烷醇基中的羟基、水解性甲硅烷基中的前述水解性基团之间进行水解缩合反应,由此可以得到固体状的聚硅氧烷化合物。
[0098]
本发明中所说的硅烷醇基是指具有与硅原子直接结合的羟基的含硅基团。本发明中所说的水解性甲硅烷基是指具有与硅原子直接结合的水解性基团的含硅基团,具体而言,例如可列举出下述通式(s-4)所示的基团。
[0099]
[化4]
[0100][0101]
(式中,r4为烷基、芳基或芳烷基等1价有机基团,r5为卤素原子、烷氧基、酰氧基、烯丙氧基、巯基、氨基、酰胺基、氨氧基、亚氨基氧基或烯氧基。
[0102]
另外,b为0~2的整数。)
[0103]
作为上述烷基,例如可列举出甲基、乙基、丙基、异丙基、丁基、异丁基、仲丁基、叔丁基、戊基、异戊基、新戊基、叔戊基、1-甲基丁基、2-甲基丁基、1,2-二甲基丙基、1-乙基丙基、己基、异己基、1-甲基戊基、2-甲基戊基、3-甲基戊基、1,1-二甲基丁基、1,2-二甲基丁基、2,2-二甲基丁基、1-乙基丁基、1,1,2-三甲基丙基、1,2,2-三甲基丙基、1-乙基-2-甲基丙基、1-乙基-1-甲基丙基等。
[0104]
作为上述芳基,例如可列举出苯基、萘基、2-甲基苯基、3-甲基苯基、4-甲基苯基、4-乙烯基苯基、3-异丙基苯基等。
[0105]
作为上述芳烷基,例如可以举出苄基、二苯基甲基、萘基甲基等。
[0106]
作为上述卤素原子,例如可列举出氟原子、氯原子、溴原子、碘原子等。
[0107]
作为上述烷氧基,例如可列举出甲氧基、乙氧基、丙氧基、异丙氧基、丁氧基、仲丁氧基、叔丁氧基等。
[0108]
作为上述酰氧基,例如可列举出甲酰氧基、乙酰氧基、丙酰氧基、丁酰氧基、新戊酰
氧基、戊酰氧基、苯基乙酰氧基、乙酰乙酰氧基、苯甲酰氧基、萘甲酰氧基等。
[0109]
作为上述烯丙氧基,例如可以举出苯氧基、萘氧基等。
[0110]
作为上述烯氧基,例如可列举出乙烯基氧基、烯丙氧基、1-丙烯氧基、异丙烯氧基、2-丁烯氧基、3-丁烯氧基、2-戊烯氧基、3-甲基-3-丁烯氧基、2-己烯氧基等。
[0111]
作为具有上述通式(s-1)和/或上述通式(s-2)所示的结构单元的聚硅氧烷链段,例如可列举出具有以下结构的聚硅氧烷链段等。
[0112]
[化5]
[0113][0114]
[化6]
[0115][0116]
[化7]
[0117][0118]
上述聚合物链段在不妨碍本发明的效果的范围内,可以根据需要具有各种官能团。作为该官能团,例如可以使用羧基、被封端的羧基、羧酸酐基、叔氨基、羟基、被封端的羟基、环碳酸酯基、环氧基、羰基、伯酰胺基、仲酰胺基、氨基甲酸酯基、下述结构式(s-5)所示的官能团等。
[0119]
[化8]
[0120][0121]
另外,上述聚合物链段可以具有乙烯基、(甲基)丙烯酰基等聚合性双键。
[0122]
本发明中使用的聚硅氧烷化合物可以通过公知的方法制造,其中优选通过下述(1)~(3)所示的方法制造。但是,不限于这些方法。
[0123]
(1)作为上述聚合物链段的原料,预先制备含有硅烷醇基和/或水解性甲硅烷基的聚合物链段,将该聚合物链段与含有兼具硅烷醇基和/或水解性甲硅烷基、以及聚合性双键的硅烷化合物的硅烷化合物混合,进行水解缩合反应的方法。
[0124]
(2)作为上述聚合物链段的原料,预先制备含有硅烷醇基和/或水解性甲硅烷基的
聚合物链段。另外,使含有兼具硅烷醇基和/或水解性甲硅烷基、以及聚合性双键的硅烷化合物的硅烷化合物进行水解缩合反应,也预先制备聚硅氧烷。然后,将聚合物链段与聚硅氧烷混合,进行水解缩合反应的方法。
[0125]
(3)将上述聚合物链段、含有兼具硅烷醇基和/或水解性甲硅烷基以及聚合性双键的硅烷化合物的硅烷化合物、与聚硅氧烷混合,进行水解缩合反应的方法。
[0126]
碳源树脂只要与聚硅氧烷化合物的混合性良好,另外,有时通过在非活性气氛中、高温烧成而被碳化,则没有特别限定,优选使用具有芳香族官能团的合成树脂类、天然化学原料,从廉价获得、杂质排除的观点出发,更优选使用酚醛树脂。
[0127]
作为合成树脂类,可列举出聚乙烯醇、聚丙烯酸等热塑性树脂、酚醛树脂、呋喃树脂等热固性树脂。作为天然化学原料,可列举出重质油,特别是作为焦油沥青类,可列举出煤焦油、焦油轻油、焦油中油、焦油重油、萘油、蒽油、煤焦油沥青、沥青油、中间相沥青、氧交联石油沥青、重油等。
[0128]
工序2中,使工序1中得到的含低氧型纳米硅粒子的浆料、聚硅氧烷化合物和碳源树脂均匀混合后,经过脱溶剂和干燥而得到混合体(前体)。原料的混合没有特别限定,可以使用具有通用的分散、混合功能的装置。其中,可以举出搅拌机、超声波混合器、预混合分散机等。在以蒸馏除去有机溶剂为目的的脱溶剂和干燥的作业中,可以使用干燥机、减压干燥机、喷雾干燥机等。
[0129]
相对于该前体的重量,优选设定硅粒子的添加量为3~50质量%、含有15~85质量%的聚硅氧烷化合物的固体成分、碳源树脂的固体成分为3~70质量%,更优选设定硅粒子的固体成分添加量为8~40质量%、聚硅氧烷化合物的固体成分为20~70质量%、碳源树脂的固体成分为3~60质量%。
[0130]
(工序3)
[0131]
工序3是通过将工序2中得到的前体在非活性气氛中进行高温烧成,使可热分解的有机成分完全分解,对其他主成分通过烧成条件的精密控制,从而得到适合于本发明的负极活性物质的烧成物的工序。具体而言,原料的聚硅氧烷化合物中存在的“si-o”键通过利用高温处理的能量进行脱水缩合反应而形成si-o-c的骨架结构,并且均匀化分散的碳源树脂也被碳化,由此在具有si-o-c骨架的三维结构体中转化为自由碳。
[0132]
工序3是将工序2中得到的前体在非活性气氛下按照烧成的程序进行烧成的工序。最高到达温度是设定的最高温度,对烧成物的结构、性能产生强烈影响。本发明中的最高到达温度优选为1020℃~1180℃,更优选为1070℃~1150℃。通过在该温度范围内进行烧成,能够使具有前述硅与碳的化学结合状态的材料的微细结构良好,还能够避免过高温烧成中的硅氧化,从而能够得到更优异的充放电特性。
[0133]
烧成方法没有特别限定,只要使用在气氛中具有加热功能的反应装置即可,可以利用连续法、分批法进行处理。关于烧成用装置,可以根据其目的适当选择流化床反应炉、回转炉、立式移动层反应炉、隧道炉、分批炉、回转窑等。
[0134]
另外,也可以在上述记载的前体烧成之前进行氧化处理。通过该氧化处理,能够在硅的表面上赋予薄的氧化膜。在用于电池时,能够防止硅表面暴露于电解液,具有抑制电解液分解的效果,从而能够提高活性物质的循环特性。关于氧化处理条件,在大气中,优选200℃~440℃的温度范围,更优选300℃~420℃。
[0135]
上述记载的非活性气氛没有特别限定,只要不含有氧化性气体即可。其中,可以使用氮、氩等,也可以使用作为还原性气氛的氮/氢的混合气体、纯氢、一氧化碳等。
[0136]
(工序4)
[0137]
工序4是将工序3中得到的烧成物粉碎,根据需要进行分级,由此得到本发明的负极活性物质的工序。粉碎可以以一个阶段进行至目标粒径,也可以分成数个阶段进行。例如烧成物为10mm以上的块或凝聚粒子、制作10μm的活性物质时,用颚式破碎机、辊式破碎机等进行粗粉碎,制成1mm左右的粒子后,用grow mill(grow磨机)、球磨机等制成100μm,用珠磨机、喷磨机等粉碎至10μm。在通过粉碎制作的粒子中有时含有粗大粒子,为了将其除去,另外在除去微粉而调整粒度分布的情况下,进行分级。所使用的分级机根据目的而分开使用风力分级机、湿式分级机等,在除去粗大粒子的情况下,通过筛的分级方式能够可靠地达成目的,因此优选。
[0138]
通过上述工序1~4的方法,得到包含在
29
si-nmr中处于-100~-110ppm的范围的峰面积(ii)相对于处于-75~-85ppm的范围的峰面积(i)之比[(ii)/(i)比]为1.0以下的低氧型纳米硅粒子的本发明的锂离子二次电池负极活性物质。通过上述制造法得到的负极活性物质的平均粒径(动态光散射法)优选为500nm~50μm,更优选为1μm~40μm,进一步优选为2~20μm。
[0139]
[锂离子二次电池用负极的制造方法]
[0140]
本发明的锂离子二次电池用负极包含上述本发明的锂离子二次电池负极活性物质。本发明的锂离子二次电池用负极通过将以上述本发明的锂离子二次电池负极活性物质和有机粘结剂为必须成分,并根据需要含有其他导电助剂等成分而构成的浆料涂布在集电体铜箔上制成薄膜而得到。本发明的锂离子二次电池负极活性物质由于包含上述低氧型纳米硅粒子,因此在将其用作负极时,发挥良好的充放电特性。
[0141]
也可以在上述浆料中加入公知惯用的石墨等碳材料来制作负极。作为该石墨等碳材料,可举出天然石墨、人工石墨、硬碳、软碳等。
[0142]
这样得到的负极由于含有本发明的负极活性物质作为活性物质,因此成为高容量且具有优异的循环特性、进而还兼具优异的首次库仑效率的二次电池用负极。该负极例如可以如下得到:将前述二次电池用负极活性物质和作为有机粘结材料的粘合剂与溶剂一起利用搅拌机、球磨机、超级砂磨机、加压捏合机等分散装置进行混炼,制备负极材料浆料,将其涂布于集电体而形成负极层。另外,也可以通过将糊状的负极材料浆料成型为片状、颗粒状等形状,将其与集电体一体化而得到。
[0143]
作为上述有机粘结剂,没有特别限定,可以列举例如:苯乙烯-丁二烯橡胶共聚物(sbr);乙烯性不饱和羧酸酯(例如(甲基)丙烯酸甲酯、(甲基)丙烯酸乙酯、(甲基)丙烯酸丁酯、(甲基)丙烯腈和(甲基)丙烯酸羟基乙酯等)和包含乙烯性不饱和羧酸(例如丙烯酸、甲基丙烯酸、衣康酸、富马酸、马来酸等)的(甲基)丙烯酸系共聚物;聚偏氟乙烯、聚环氧乙烷、聚环氧氯丙烷、聚磷腈、聚丙烯腈、聚酰亚胺、聚酰胺酰亚胺、羧甲基纤维素(cmc)等高分子化合物。作为上述有机粘结剂,也可以采用化学稳定性高的水性粘合剂。
[0144]
这些有机粘结剂根据各自的物性,有分散或溶解于水中而得到的有机粘结剂,另外,有溶解于n-甲基-2-吡咯烷酮(nmp)等有机溶剂中而得到的有机粘结剂。锂离子二次电池负极的负极层中的有机粘结剂的含有比率优选为1~30质量%,更优选为2~20质量%,
进一步优选为3~15质量%。
[0145]
通过使有机粘结剂的含有比率为1质量%以上,密合性良好,由于充放电时的膨胀、收缩而造成的负极结构的破坏得到抑制。另一方面,通过为30质量%以下,可抑制电极电阻的上升。
[0146]
另外,在上述负极材料浆料中,根据需要可以混合导电助剂。作为导电助剂,例如可列举出炭黑、石墨、乙炔黑、或者显示导电性的氧化物、氮化物等。导电助剂的使用量相对于本发明的负极活性物质为1~15质量%左右即可。
[0147]
上述集电体的材质和形状没有特别限定,可以使用将铜、镍、钛、不锈钢等制成箔状、开孔箔状、网状等而得到的带状集电体。另外,也可以使用多孔性材料,例如多孔金属(发泡金属)、碳纸等。
[0148]
作为将上述负极材料浆料涂布于集电体的方法,没有特别限定,例如可列举出金属掩模印刷法、静电涂装法、浸涂法、喷涂法、辊涂法、刮刀法、凹版涂布法、丝网印刷法等公知的方法。涂布后,优选根据需要利用平板压机、压延辊等进行压延处理。
[0149]
另外,成型为片状、颗粒状等形状的负极材料浆料与集电体的一体化例如可以通过辊、压制或它们的组合等公知的方法来进行。
[0150]
形成于上述集电体上的负极层和与集电体一体化的负极层优选根据所使用的有机粘结剂进行热处理。例如,在使用公知惯用的水体系的苯乙烯-丁二烯橡胶共聚物(sbr)等的情况下,在100~130℃进行热处理即可,在使用以聚酰亚胺、聚酰胺酰亚胺为主骨架的有机粘结剂的情况下,优选在150~450℃进行热处理。
[0151]
通过该热处理,由溶剂的除去、粘合剂的固化带来的高强度化得到发展,能够提高粒子间以及粒子与集电体间的密合性。需要说明的是,为了防止处理中的集电体的氧化,这些热处理优选在氦、氩、氮等非活性气氛、真空气氛下进行。
[0152]
另外,在进行热处理之后,负极优选预先进行压制(加压处理)。在使用了本发明的负极活性物质的二次电池用负极中,电极密度优选为1.0~1.8g/cm3,更优选为1.1~1.7g/cm3,进一步优选为1.2~1.6g/cm3。关于电极密度,越高则越有密合性及电极的体积容量密度提高的倾向,但若密度过高,则电极中的空隙减少,由此硅等的体积膨胀抑制效果变弱,因此循环特性降低。
[0153]
[锂离子二次电池(全电池)的构成]
[0154]
如上所述,使用了本发明的锂离子二次电池负极活性物质的负极由于充放电特性优异,因此只要是二次电池就没有特别限定,优选用于非水电解质二次电池和固体型电解质二次电池,特别是在用作非水电解质二次电池的负极时发挥优异的性能。
[0155]
本发明的锂离子二次电池的特征在于,使用含有上述本发明的锂离子二次电池负极活性物质的锂离子二次电池用负极而成。例如,在用于湿式电解质二次电池的情况下,可以通过将正极和本发明的负极隔着隔膜相对配置,注入电解液来构成。通过按照该构成来组装二次电池,可以制造本发明的锂离子二次电池。
[0156]
上述正极可以与上述负极同样地通过在集电体表面上形成正极层而得到。此时的集电体可以使用将铝、钛、不锈钢等金属、合金制成箔状、开孔箔状、网状等而成的带状集电体。
[0157]
作为上述正极层中使用的正极材料,没有特别限制。在非水电解质二次电池中,在
制作锂离子二次电池的情况下,例如使用能够掺杂或嵌入锂离子的金属化合物、金属氧化物、金属硫化物、或导电性高分子材料即可,没有特别限定。例如,可以将钴酸锂(licoo2)、镍酸锂(linio2)、锰酸锂(limno2)、以及它们的复合氧化物(licoxniymnzo2、x y z=1)、锂锰尖晶石(limn2o4)、锂钒化合物、v2o5、v6o
13
、vo2、mno2、tio2、mov2o8、tis2、v2s5、vs2、mos2、mos3、cr3o8、cr2o5、橄榄石型limpo4(m:co、ni、mn、fe)、聚乙炔、聚苯胺、聚吡咯、聚噻吩、多并苯等导电性聚合物、多孔质碳等单独使用或混合使用。
[0158]
作为上述隔膜,例如可以使用以聚乙烯、聚丙烯等聚烯烃为主要成分的无纺布、布、微孔膜或它们的组合。需要说明的是,在制成所制作的非水电解质二次电池的正极与负极不直接接触的结构的情况下,不需要使用隔膜。
[0159]
作为上述电解液,例如可以使用将liclo4、lipf6、liasf6、libf4、liso3cf3等锂盐溶解于碳酸亚乙酯、碳酸亚丙酯、碳酸亚丁酯、碳酸亚乙烯酯、氟代碳酸亚乙酯、环戊酮、环丁砜、3-甲基环丁砜、2,4-二甲基环丁砜、3-甲基-1,3-噁唑烷-2-酮、γ-丁内酯、碳酸二甲酯、碳酸二乙酯、碳酸乙基甲酯、碳酸甲基丙酯、碳酸丁基甲酯、碳酸乙基丙酯、碳酸丁基乙酯、碳酸二丙酯、1,2-二甲氧基乙烷、四氢呋喃、2-甲基四氢呋喃、1,3-二氧戊环、乙酸甲酯、乙酸乙酯等单体或2个成分以上的混合物的非水系溶剂中而得的所谓的有机电解液。
[0160]
本发明的锂离子二次电池的结构没有特别限定,通常,将正极和负极以及根据需要设置的隔膜卷绕成扁平漩涡状而制成卷绕式极板组,或者将它们以平板状的方式层叠而制成层叠式极板组,通常制成将这些极板组封入外装体中的结构。
[0161]
本发明的锂离子二次电池没有特别限定,可以作为纸型电池、纽扣型电池、硬币型电池、层叠型电池、圆筒型电池、方型电池等使用。上述本发明的锂离子二次电池负极活性物质也可以应用于以嵌入脱嵌锂离子为充放电机制的所有电化学装置,例如混合电容器、固体锂二次电池等。
[0162]
实施例
[0163]
利用以下的实施例1-4及比较例1-2中记载的方法制作含硅粒子浆料,进而利用实施例5-8及比较例3的方法制作半电池。添加剂及硅粒子浆料的物性如下述表1所示,电池特性如下述表2所示。
[0164]
(实施例1:含硅粒子浆料的制作)
[0165]
将70g市售品单质硅粉末(高纯度化学制、纯度99.9%、平均粒径2-4μm)、28g添加剂1、392g mek混合,充分搅拌。使用株式会社广岛金属&机械公司制ultra-apex mill uam-015对该混合液进行6小时湿式粉碎处理,得到含有平均粒径(d50)为53.9nm的硅粒子、粘度为1.84mpa
·
s、110℃时的不挥发成分为20.0%的浆料。得到的浆料为纳米化的硅粒子均匀地分散在分散介质中的状态(纳米化“〇”)。平均粒径(d50)和粘度的测定方法如下所述。另外,通过以下的“29
si-nmr的测定及分析方法”进行的硅粒子的
29
si-nmr中的处于-100~~110ppm的范围的峰面积(ii)相对于处于-75~~85ppm的范围的峰面积(i)之比[(ii)/(i)比]为0.43。表1表示浆料物性,图1表示
29
si-nmr光谱的图表,图2表示硅粒子的透射型电子显微镜(tem)图像。
[0166]
(实施例2:含硅粒子浆料的制作)
[0167]
除了使用添加剂2代替添加剂1以外,与实施例1同样地进行浆料的制作,得到含有平均粒径(d50)为44.9nm的硅粒子、粘度为2.40mpa
·
s、110℃时的不挥发成分为20.3%的
浆料。浆料中的硅粒子的
29
si-nmr峰面积比[(ii)/(i)比]为0.23。表1表示浆料物性,图3表示
29
si-nmr光谱的图表。
[0168]
(实施例3:含硅粒子浆料的制作)
[0169]
除了使用添加剂3代替添加剂1以外,与实施例1同样地进行浆料的制作,得到含有平均粒径(d50)为62.0nm的硅粒子、粘度为1.97mpa
·
s、110℃时的不挥发成分为20.1%的浆料。浆料中的硅粒子的
29
si-nmr峰面积比[(ii)/(i)比]为0.76。表1表示浆料物性,图4表示
29
si-nmr光谱的图表,图5表示硅粒子的透射型电子显微镜(tem)图像。
[0170]
(实施例4:含硅粒子浆料的制作)
[0171]
除了并用添加剂1和添加剂2代替添加剂1以外,与实施例1同样地进行浆料的制作,得到含有平均粒径(d50)为53.9nm的硅粒子、粘度为1.59mpa
·
s、110℃时的不挥发成分为20.5%的浆料。浆料中的硅粒子的
29
si-nmr峰面积比[(ii)/(i)比]为0.05。表1表示浆料物性,图6表示
29
si-nmr光谱的图表。
[0172]
(实施例5:含硅粒子浆料的制作)
[0173]
使用添加剂1,使添加剂量为5份,除此以外,与实施例1同样地进行浆料的制作,得到含有平均粒径(d50)为83.1nm的硅粒子、粘度为9.21mpa
·
s、110℃时的不挥发成分为16.6%的浆料。浆料中的硅粒子的
29
si-nmr峰面积比[(ii)/(i)比]为0.73。
[0174]
(实施例6:含硅粒子浆料的制作)
[0175]
使用添加剂1,使添加剂量为20份,除此以外,与实施例1同样地进行浆料的制作,得到含有平均粒径(d50)为57.8nm的硅粒子、粘度为3.32mpa
·
s、110℃时的不挥发成分为18.3%的浆料。浆料中的硅粒子的
29
si-nmr峰面积比[(ii)/(i)比]为0.56。
[0176]
(实施例7:含硅粒子浆料的制作)
[0177]
使用添加剂1,使添加剂量为60份,除此以外,与实施例1同样地进行浆料的制作,得到含有平均粒径(d50)为59.2nm的硅粒子、粘度为1.45mpa
·
s、110℃时的不挥发成分为22.8%的浆料。浆料中的硅粒子的
29
si-nmr峰面积比[(ii)/(i)比]为0.40。
[0178]
(实施例8:含硅粒子浆料的制作)
[0179]
使用添加剂1,使添加剂量为80份,使粉碎处理时间为8小时,除此以外,与实施例1同样地进行浆料的制作,得到含有平均粒径(d50)为82.7nm的硅粒子、粘度为1.38mpa
·
s、110℃时的不挥发成分为24.9%的浆料。浆料中的硅粒子的
29
si-nmr峰面积比[(ii)/(i)比]为0.45。
[0180]
(实施例9:含硅粒子浆料的制作)
[0181]
除了使用添加剂5代替添加剂1以外,与实施例1同样地进行浆料的制作,得到含有平均粒径(d50)为90.4nm的硅粒子、粘度为3.96mpa
·
s、110℃时的不挥发成分为20.5%的浆料。浆料中的硅粒子的
29
si-nmr峰面积比[(ii)/(i)比]为0.65。
[0182]
(实施例10:含硅粒子浆料的制作)
[0183]
除了使用添加剂6代替添加剂1以外,与实施例1同样地进行浆料的制作,得到含有平均粒径(d50)为79.2nm的硅粒子、粘度为5.93mpa
·
s、110℃时的不挥发成分为20.1%的浆料。浆料中的硅粒子的
29
si-nmr峰面积比[(ii)/(i)比]为0.38。
[0184]
(实施例11:含硅粒子浆料的制作)
[0185]
除了使用添加剂7代替添加剂1以外,与实施例1同样地进行浆料的制作,得到含有
平均粒径(d50)为75.4nm的硅粒子、粘度为1.83mpa
·
s、110℃时的不挥发成分为20.3%的浆料。浆料中的硅粒子的
29
si-nmr峰面积比[(ii)/(i)比]为0.66。
[0186]
(实施例12:含硅粒子浆料的制作)
[0187]
除了使用添加剂8代替添加剂1以外,与实施例1同样地进行浆料的制作,得到含有平均粒径(d50)为81.1nm的硅粒子、粘度为2.90mpa
·
s、110℃时的不挥发成分为20.3%的浆料。浆料中的硅粒子的
29
si-nmr峰面积比[(ii)/(i)比]为0.81。
[0188]
(实施例13:含硅粒子浆料的制作)
[0189]
除了设为98g市售品单质硅粉末(高纯度化学制、纯度99.9%、平均粒径2-4μm)、39.2g添加剂1、244g mek以外,与实施例1同样地进行浆料的制作。得到含有平均粒径(d50)为76.0nm的硅粒子、粘度为6.52mpa
·
s、110℃时的不挥发成分为36.02%的浆料。浆料中的硅粒子的
29
si-nmr峰面积比[(ii)/(i)比]为0.53。
[0190]
(实施例14:含硅粒子浆料的制作)
[0191]
除了设为112g市售品单质硅粉末(高纯度化学制、纯度99.9%、平均粒径2-4μm)、44.8g添加剂1、235g mek以外,与实施例1同样地进行浆料的制作。得到含有平均粒径(d50)为79.2nm的硅粒子、粘度为7.22mpa
·
s、110℃时的不挥发成分为40.10%的浆料。浆料中的硅粒子的
29
si-nmr峰面积比[(ii)/(i)比]为0.51。
[0192]
(比较例1:含硅粒子浆料的制作)
[0193]
除了使用添加剂4代替添加剂1以外,与实施例1同样地进行浆料的制作,得到含有平均粒径(d50)为57.9nm的硅粒子、粘度为13.4mpa
·
s、110℃时的不挥发成分为20.2%的浆料。浆料中的硅粒子的
29
si-nmr峰面积比[(ii)/(i)比]为1.20。表1中示出浆料物性,图7中示出硅粒子的透射型电子显微镜(tem)图像。
[0194]
(比较例2:含硅粒子浆料的制作)
[0195]
除了不使用添加剂以外,与实施例1同样地进行浆料的制作,得到含有平均粒径(d50)为500nm以上的硅粒子、粘度为30.0mpa
·
s以上、110℃时的不挥发成分为20.0%的浆料。得到的浆料中,硅粒子粗大,为对于分散介质而言没有分散性的状态(纳米化
“×”
)。表1中示出浆料物性。
[0196]
[表1]
[0197][0198]
※
添加剂量表示相对于硅粉末100质量份所使用的添加剂的比例(质量份)。
[0199]
(含硅粒子浆料的粘度测定方法)
[0200]
对于含有硅粒子的浆料,使用锥板型粘度计(brookfield公司制),在23℃、100rpm测定粘度。
[0201]
(硅粒子平均粒径测定方法)
[0202]
将浆料用mek稀释后,利用超声波进行1分钟分散处理,利用激光粒度分析仪(laser microsizer lms-3000、seishin公司制),使用mek作为分散溶剂测定含硅粒子的浆料。
[0203]
(
29
si-nmr的测定及分析方法)
[0204]
将样品采集到固体nmr试样管中,利用固体nmr分析装置(jeol resonace制、jnm-eca600)进行单脉冲测定。对于利用delta5对所获得的固体nmr光谱数据进行傅立叶变换,对由此所得的数据,利用acd labs软件,使用高斯(gauss) 洛伦兹(lorentz)函数进行波形分离。基于通过波形分离得到的峰面积,算出处于-100~-110ppm的范围的峰面积(ii)相对于处于-75~-85ppm的范围的峰面积(i)的比[(ii)/(i)比]。
[0205]
(实施例15)
[0206]
将实施例1中得到的含硅浆料、聚硅氧烷化合物和甲阶型酚醛树脂以一定的构成比(以烧成后的组成计算的投料构成:sioc/c/si=10/35/55)均匀混合后,投入3口可拆式烧瓶中。盖住1个口,在2个口连接氮导入管、溶剂捕获装置。向烧瓶内导入氮,一边用磁力搅
拌器搅拌混合液,一边在油浴中将烧瓶加热至120℃,蒸馏除去溶剂直至搅拌子不移动。然后,冷却至室温,得到作为烧成前体的树脂干燥物。
[0207]
将得到的前体在氮气氛中以1050℃、6小时进行高温烧成,由此得到黑色固体物。将得到的黑色固体物用行星式球磨机以乙醇为溶剂进行湿式粉碎,粉碎后除去溶剂并干燥,得到黑色粉末活性物质粒子。
[0208]
如以下的“半电池的制作及充放电特性的测定”中记载的那样,制作使用了所得到的黑色粉末活性物质粒子的半电池并测定充放电特性,结果放电容量为1461mah/g,库仑效率为80.2%,10个循环的容量维持率为90.2%。表2中示出电池特性。
[0209]
(实施例16)
[0210]
使用实施例2中得到的含硅浆料代替实施例1中得到的含硅浆料,除此以外,与实施例13同样地制作黑色粉末活性物质粒子,得到半电池。测定充放电特性,结果放电容量为1451mah/g,库仑效率为79.0%,10个循环的容量维持率为89.8%。表2中示出电池特性。
[0211]
(实施例17)
[0212]
使用实施例3中得到的含硅浆料代替实施例1中得到的含硅浆料,除此以外,与实施例13同样地制作黑色粉末活性物质粒子,得到半电池。测定充放电特性,结果放电容量为1075mah/g,库仑效率为75.0%,10个循环的容量维持率为92.7%。表2中示出电池特性。
[0213]
(实施例18)
[0214]
使用实施例4中得到的含硅浆料代替实施例1中得到的含硅浆料,除此以外,与实施例13同样地制作黑色粉末活性物质粒子,得到半电池。测定充放电特性,结果放电容量为1561mah/g,库仑效率为81.2%,10个循环的容量维持率为86.4%。表2中示出电池特性。
[0215]
(实施例19)
[0216]
使用实施例5中得到的含硅浆料代替实施例1中得到的含硅浆料,除此以外,与实施例13同样地制作黑色粉末活性物质粒子,得到半电池。测定充放电特性,结果放电容量为1204mah/g,库仑效率为80.2%,10个循环的容量维持率为91.4%。表2中示出电池特性。
[0217]
(实施例20)
[0218]
使用实施例6中得到的含硅浆料代替实施例1中得到的含硅浆料,除此以外,与实施例13同样地制作黑色粉末活性物质粒子,得到半电池。测定充放电特性,结果放电容量为1415mah/g,库仑效率为80.3%,10个循环的容量维持率为89.9%。表2中示出电池特性。
[0219]
(实施例21)
[0220]
使用实施例7中得到的含硅浆料代替实施例1中得到的含硅浆料,除此以外,与实施例13同样地制作黑色粉末活性物质粒子,得到半电池。测定充放电特性,结果放电容量为1453mah/g,库仑效率为80.1%,10个循环的容量维持率为90.1%。表2中示出电池特性。
[0221]
(实施例22)
[0222]
使用实施例8中得到的含硅浆料代替实施例1中得到的含硅浆料,除此以外,与实施例13同样地制作黑色粉末活性物质粒子,得到半电池。测定充放电特性,结果放电容量为1464mah/g,库仑效率为80.0%,10个循环的容量维持率为89.7%。表2中示出电池特性。
[0223]
(实施例23)
[0224]
使用实施例9中得到的含硅浆料代替实施例1中得到的含硅浆料,除此以外,与实施例13同样地制作黑色粉末活性物质粒子,得到半电池。测定充放电特性,结果放电容量为
1487mah/g,库仑效率为80.7%,10个循环的容量维持率为88.2%。表2中示出电池特性。
[0225]
(实施例24)
[0226]
使用实施例10中得到的含硅浆料代替实施例1中得到的含硅浆料,除此以外,与实施例13同样地制作黑色粉末活性物质粒子,得到半电池。测定充放电特性,结果放电容量为1474mah/g,库仑效率为80.3%,10个循环的容量维持率为89.3%。表2中示出电池特性。
[0227]
(实施例25)
[0228]
使用实施例11中所获得的含硅浆料代替实施例1中所获得的含硅浆料,除此以外,与实施例13同样地制作黑色粉末活性物质粒子,获得半电池。测定充放电特性,结果放电容量为1442mah/g,库仑效率为79.2%,10个循环的容量维持率为88.9%。表2中示出电池特性。
[0229]
(实施例26)
[0230]
使用实施例12中得到的含硅浆料代替实施例1中得到的含硅浆料,除此以外,与实施例13同样地制作黑色粉末活性物质粒子,得到半电池。测定充放电特性,结果放电容量为1449mah/g,库仑效率为80.5%,10个循环的容量维持率为89.3%。表2中示出电池特性。
[0231]
(实施例27)
[0232]
使用实施例13中得到的含硅浆料代替实施例1中得到的含硅浆料,除此以外,与实施例13同样地制作黑色粉末活性物质粒子,得到半电池。测定充放电特性,结果放电容量为1460mah/g,库仑效率为81.0%,10个循环的容量维持率为88.7%。表2中示出电池特性。
[0233]
(实施例28)
[0234]
使用实施例13中得到的含硅浆料代替实施例1中得到的含硅浆料,除此以外,与实施例13同样地制作黑色粉末活性物质粒子,得到半电池。测定充放电特性,结果放电容量为1452mah/g,库仑效率为80.6%,10个循环的容量维持率为89.1%。表2中示出电池特性。
[0235]
(比较例3)
[0236]
使用比较例1中得到的含硅浆料代替实施例1中得到的含硅浆料,除此以外,与实施例5同样地制作黑色粉末活性物质粒子,得到半电池。测定充放电特性,结果放电容量为551mah/g,库仑效率为62.0%,10个循环的容量维持率为97.8%。表2中示出电池特性。
[0237]
[表2]
[0238][0239]
(半电池的制作和充放电特性的测定)
[0240]
如下述那样组装使用了本发明的负极活性物质的评价用半电池,测定充放电特性。
[0241]
将负极活性物质(8份)、导电助剂乙炔黑(1份)和有机粘结剂(1份、明细:市售品sbr(0.75份) cmc(0.25份)和蒸馏水(10份)混合,用自转公转式的脱泡搅拌机(泡取
り
錬太郎)搅拌10分钟,由此制备浆料。使用敷料器在厚度20μm的铜箔上涂膜后,在110℃、减压条件下干燥,得到厚度约40μm的电极薄膜。冲裁成直径14mm的圆状电极,在20mpa的压力下压制。在氧浓度低(《10ppm)和水分含量极低(露点-40
°
以下)的手套箱中,以li箔作为对电极,隔着25μm的聚丙烯制隔膜使本发明的电极相对,吸附电解液(岸田化学、1mol/l的lipf6、碳酸二乙酯∶碳酸亚乙酯=1∶1(容积比)),制作评价用半电池(cr2032型)。
[0242]
使用二次电池充放电试验装置(北斗电工)测定电池特性,在室温25℃、截止电压范围为0.005~1.5v、充放电速率为0.1c(1~3次)和0.2c(4个循环以后)的条件下,在定电流-定电压式充电/定电流式放电的设定条件下进行充放电特性的评价试验。在各充放电时的切换时,以开路放置30分钟。首次库仑效率和循环特性(本技术中是指10个循环时的容量维持率)如下求出。
[0243]
首次库伦效率(%)=首次放电容量(mah/g)/首次充电容量(mah/g)
[0244]
容量维持率(第十次)=第十次的放电容量(mah/g)/首次放电容量(mah/g)
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。