1.本发明涉及生物医药领域,具体地涉及一种可注射透明质酸微球及其制备方法以及在例如组织填充等领域中的用途。
背景技术:
2.随着人类年龄增长或受某些疾病影响,人体中肌肉和胶原蛋白组织会产生不同程度的功能性退化,造成皮肤凹陷、胃液反流等问题,因此人们发明了多种填充剂,以此来填补凹陷皮肤或通过异物刺激肌肉和胶原蛋白的再生长,如玻尿酸、牛胶原蛋白等。但这些物质作为填充剂所起到的填充效果维持时间较短,需要频繁在注射保持其填充效果。
3.为达到长期填充效果,人们又尝试使用生物的不可降解的材料制作成微球作为填充剂,如聚乙烯醇(pva)、聚甲基丙基酸甲酯(pmma)等。尽管这些填充剂的填充效果维持时间明显增长,但这些材料在体内留存过久,会释放有害物质,并因此引发一系列的副反应,危害人体健康。
4.近年来,生物可降解的高分子材料走入人们的视野,该类型材料对人体无毒、无排斥反应,并可以随着人体代谢逐渐降解后排出体外。
5.聚羟基脂肪酸酯,英文名为polyhydroxyalkanoates,简称pha,是一种天然的高分子生物材料,由微生物合成的一种细胞内聚酯。由于pha具有良好的生物相容性能、生物可降解性,是当下最为理想的生物医学材料之一。
6.现有技术中,在将pha材料制备可注射微球时,容易遇到制得的微球十分容易团聚、粘连,形成较大的块状物质的问题,给后续注射带来不便,并且所制备微球粘附细胞的能力不强,不能很好地与组织相容,极大的阻碍了生物可降解材料微球作为填充剂的应用。因此,急需一种新型的微球制备方案,解决微球的细胞粘附性不高、易团聚、粘连以及组织相容性不足等的问题。
7.背景技术中的信息仅仅在于说明本发明的总体背景,不应视为承认或以任何形式暗示这些信息构成本领域一般技术人员所公知的现有技术。
技术实现要素:
8.为解决现有技术中的至少部分技术问题,本发明提供一种可注射透明质酸微球,在相同条件下,本发明的微球不易团聚和粘连,具有更高的细胞粘附性和组织相容性。具体地,本发明包括以下内容。
9.本发明的一方面,提供一种可注射透明质酸微球的制备方法,其包括以下步骤:
10.(1)将交联透明质酸加入水性溶剂中制备第一液相,所述第一液相中交联透明质酸的浓度控制在基于重量为0.1-5%;
11.(2)将内聚酯加入有机溶剂中制备第二液相,所述第二液相中内聚酯的浓度控制为0.1-500mg/ml;和
12.(3)将所述第一液相与所述第二液相以3-15:1的体积比混合制成微球。
13.根据本发明所述的可注射透明质酸微球的制备方法,优选地,所述交联透明质酸的交联度为5%-50%。
14.根据本发明所述的可注射透明质酸微球的制备方法,优选地,所述交联透明质酸的均重分子量为9千-2千万道尔。
15.根据本发明所述的可注射透明质酸微球的制备方法,优选地,所述内聚酯的重均分子量为2-100万道尔,且选自聚(3-羟基丁酸-co-3-羟基戊酸-co-3-羟基己酸)、聚(3-羟基丁酸-3-羟基己酸)、聚(3-羟基丁酸-4-羟基丁酸)和聚(3-羟基丁酸-co-3-羟基戊酸)中的一种或多种。
16.根据本发明所述的可注射透明质酸微球的制备方法,优选地,所述微球的粒径在20-60μm。
17.根据本发明所述的可注射透明质酸微球的制备方法,优选地,所述步骤(1)中的水性溶剂为水,所述步骤(2)中的有机溶剂选自n-甲基吡咯烷酮、二氯甲烷、三氯甲烷和乙腈中的一种或多种。
18.本发明的另一方面,提供一种可注射透明质酸微球,其通过根据第一方面所述的方法制备得到。
19.优选地,本发明的可注射透明质酸微球的杨氏模量为1000pa以上。
20.根据本发明所述的可注射透明质酸微球,优选地,其具有延长的降解时间。
21.本发明的又一方面,提供可注射透明质酸微球在制备组织填充剂中的用途。
22.本发明的可注射透明质酸微球在相同条件下具有更高的杨氏模量,达到1000pa以上,并且具有延长的降解时间,不易团聚和粘连,具有更高的细胞粘附性和组织相容性。
附图说明
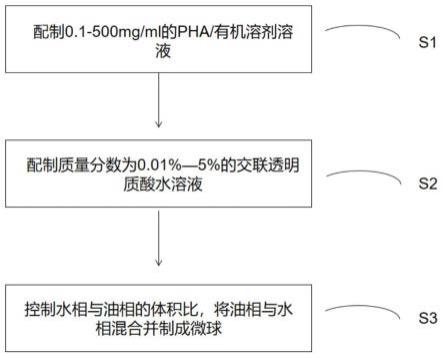
23.图1本发明示例性制备方法的流程图。
24.图2本发明实施例1制得的微球的电镜图。
25.图3用激光粒度仪进行粒径和span值的测试本发明实施例1制得的微球的结果。
26.图4牛血清白蛋白吸附实验标准曲线。
具体实施方式
27.现详细说明本发明的多种示例性实施方式,该详细说明不应认为是对本发明的限制,而应理解为是对本发明的某些方面、特性和实施方案的更详细的描述。
28.应理解本发明中所述的术语仅仅是为描述特别的实施方式,并非用于限制本发明。另外,对于本发明中的数值范围,应理解为具体公开了该范围的上限和下限以及它们之间的每个中间值。在任何陈述值或陈述范围内的中间值以及任何其他陈述值或在所述范围内的中间值之间的每个较小的范围也包括在本发明内。这些较小范围的上限和下限可独立地包括或排除在范围内。
29.除非另有说明,否则本文使用的所有技术和科学术语具有本发明所述领域的常规技术人员通常理解的相同含义。虽然本发明仅描述了优选的方法和材料,但是在本发明的实施或测试中也可以使用与本文所述相似或等同的任何方法和材料。本说明书中提到的所有文献通过引用并入,用以公开和描述与所述文献相关的方法和/或材料。在与任何并入的
文献冲突时,以本说明书的内容为准。除非另有说明,否则“%”为基于重量的百分数。
30.[可注射透明质酸微球]
[0031]
本发明的一方面,提供一种可注射透明质酸微球,本文有时可简称为“本发明的微球”。本发明的微球不易团聚和粘连,但具有更高的细胞粘附性和组织相容性。
[0032]
本发明的微球包括外层亲水层和内部疏水层。其中,亲水层为交联透明质酸层,疏水层为内聚酯类化合物,通过使含有交联透明质酸的水相包覆含有内聚酯类化合物的油相制备得到。
[0033]
本发明中使用交联透明质酸,而不是未交联透明质酸。本发明中通过交联使透明质酸分子更加团聚,加强了透明质酸的支撑力,使透明质酸成为良好的填充材料。与未交联透明质酸相比,交联透明质酸还大大提升了细胞的粘附性和组织相容性。
[0034]
本发明中交联透明质酸的交联度不特别特定,但一般控制在5-55%,优选10-50%,更优选20-40%。如果交联度不在上述范围内,则对于本发明的效果而言是不利的。一方面,当交联度变低,例如低于5%时,则细胞粘附性变弱,并且杨氏模量变低。另一方面,当交联度变高,例如高于55%时,则制备的微球倾向于不稳定,甚至出现破裂。交联度可通过例如交联剂的使用量和反应时间等来进行控制。
[0035]
本发明中交联透明质酸的分子量不特别限定,一般可控制在0.9万-2千万道尔之间,优选1万-1000万道尔之间,更优选5万-500万道尔之间。分子量越大,则越不易被降解,但如果分子量过大,则细胞粘附性变差。
[0036]
本发明中内聚酯类化合物是指微生物体存在的天然高分子生物材料,其在体内与细胞具有良好的细胞相容性。内聚酯类化合物的类型不特别限定,其实例包括但不限于聚(3-羟基丁酸-co-3-羟基戊酸-co-3-羟基己酸)、聚(3-羟基丁酸-3-羟基己酸)、聚(3-羟基丁酸-4-羟基丁酸)和聚(3-羟基丁酸-co-3-羟基戊酸)。本发明可以使用上述成分中的一种或多种。在使用多种成分的情况下,各成分之间的用量比不特别限定,可根据需要由本领域技术人员自由配比。
[0037]
本发明中内聚酯类化合物的分子量不特别限定,但一般控制在2-100万道尔之间,优选5-90万道尔之间,更优选10-80万道尔之间,例如,20万、30万、40万、50万、60万、70万等。
[0038]
本发明中微球的粒径需控制在20-60μm之间,优选30-50μm之间。20-60μm的粒径对于本发明的目的而言是必要的。如果微球直径在20微米以下,则可能会被人体细胞所吞噬。另一方面,如果微球过大,则不利于注射,严重时可能会堵塞针头,甚至引起皮肤破裂。虽然微球的粒径可以在上述范围内变化,但本发明的微球具有更加均一的粒径分布。
[0039]
本发明的微球在相同条件下具有更高的杨氏模量。优选地本发明的微球的杨氏模量为1000pa以上,例如1100pa以上、1200pa以上、1300pa以上等。
[0040]
[制备方法]
[0041]
本发明的另一方面,提供一种可注射透明质酸微球的制备方法,其包括但不限于以下步骤:
[0042]
(1)将交联透明质酸加入水性溶剂中制备第一液相,所述第一液相中交联透明质酸的浓度控制在基于重量为0.1-5%;
[0043]
(2)将内聚酯加入有机溶剂中制备第二液相,所述第二液相中内聚酯的浓度控制
为0.1-500mg/ml;和
[0044]
(3)将所述第一液相与所述第二液相以3-15:1的体积比混合制成微球。
[0045]
本发明中,步骤(1)为制备第一液相的步骤,第一液相是水相。步骤(1)包括将交联透明质酸溶解或分散于水性溶剂。其中水性溶剂包括水以及其与其他水性溶剂的混合物。本发明优选使用水,例如纯净水或蒸馏水等。在溶解或分散时可通过例如搅拌来加速。本发明的第一液相中交联透明质酸的浓度一般控制在基于重量为0.1-5%,优选0.5-4%,更优选1-3%,例如1%、1.5%、2%、2.5%和3%等。
[0046]
本发明中,步骤(2)为制备第二液相的步骤,第二液相为油相。步骤(2)包括将所需量的内聚酯溶解于有机溶剂中,配制一定浓度溶液。其中,有机溶剂的实例包括n-甲基吡咯烷酮、二氯甲烷、三氯甲烷和乙腈。本发明可以使用上述溶剂中的一种或多种。在使用多种溶剂的情况下,各溶剂的用量比不特别限定,可以根据需要由技术人员自由设定。在示例性步骤(2)中包括将0.8g的p34hb溶于20ml的二氯甲烷中,配制40mg/ml的pha/有机溶剂溶液。
[0047]
本发明中,步骤(3)是两相混合制备微球的步骤。第一液相的体积需大于第二液相的体积,两者的体积比一般在(3-15):1范围内,优选4-10:1范围内,更优选5-8:1范围内。第一液相的体积相对过小或过大均不利于微球形成。例如,如果第一液相与第二液相的体积相等,或小于第二液相,则不会形成本发明的透明质酸微球。
[0048]
本发明中,第一液相与第二液相的混合可通过膜乳化器进行。在使用膜乳化器进行混合时控制乳化压力在0.01-0.1mpa之间,优选0.02-0.05mpa之间。混合乳化时间一般在60-240分钟之间,优选90-200分钟之间,更优选100-150分钟之间。混合乳化时的搅拌转速可控制在100-500r/min,优选150-400r/min,更优选200-350r/min。乳化完成后可进一步进行固化。
[0049]
在示例性混合乳化方案中,控制第二液相与第一液相的体积比为1/5,乳化压力0.04mpa,膜管长度为15μm,乳化时长为120min;磁子长度为5cm,乳化搅拌转速为350r/min。乳化完成后,开始固化,固化使用顶置搅拌器、10cm搅拌桨,另加入280ml水,搅拌转速为120r/min,固化时长24h,得到可注射pha微球。
[0050]
本领域技术人员应理解,只要能够实现本发明的目的,步骤(1)和(2)的顺序并不特别限定。此外,两个步骤可同时进行。另外,本领域技术人员还应理解的是,在上述步骤(1)-(3)前后,或这些任意步骤之间还可包含其他步骤或操作,例如进一步优化和/或改善本发明所述的方法。
[0051]
[用途]
[0052]
本发明还提供可注射透明质酸微球的用途,优选用于制备组织填充剂。本发明的微球具有良好的生物相容性能、生物可降解性,在体内与细胞具有良好的细胞相容性,细胞可以在此种微球上良好生长,且该种微球可以降解为co2和h2o,不会生成有害成分。
[0053]
实施例1
[0054]
本实施例的制备方法如下所示:
[0055]
s1、制备油相:将0.8g的p34hb溶于20ml的二氯甲烷中,配制40mg/ml的pha/有机溶剂溶液,其中,p34hb的重均分子量为15万;
[0056]
s2、制备水相:配制质量分数为3%的交联透明质酸(交联度30%)水溶液;
[0057]
s3、将油相通过膜乳化器乳化进入转动的水相中,控制油相与水相的体积比为1/
5,乳化压力0.04mpa,膜管长度为15μm,乳化时长为120min;磁子长度为5cm,乳化搅拌转速为350r/min。乳化完成后,开始固化,固化使用顶置搅拌器、10cm搅拌桨,另加入280ml水,搅拌转速为120r/min,固化时长24h,即得到可注射pha微球。
[0058]
实施例2
[0059]
本实施例的制备方法如下所示:
[0060]
s1、制备油相:将100mg的p34hb溶于20ml的三氯甲烷中,配制500mg/ml的pha/有机溶剂溶液,其中,p34hb的重均分子量为10万;
[0061]
s2、制备水相:配制质量分数为4%的交联透明质酸(交联度40%)水溶液;
[0062]
s3、控制油相与水相的体积比为1/3,将油相与水相混合,通过常规的乳化挥发法制成微球,具体为:先将油水混合液用磁力搅拌,转速为300r/min,搅拌5min,然后降低转速至150r/min,搅拌24h,使有机溶剂三氯甲烷挥发,最后将混合液离心,收集清洗微球,进行冻干,即可得到可注射p34hb微球。
[0063]
比较例1
[0064]
s1、制备油相:将0.8g的p34hb溶于20ml的二氯甲烷中,配制40mg/ml的pha/有机溶剂溶液,其中,p34hb的重均分子量为15万;
[0065]
s2、制备水相:配制质量分数为3%透明质酸(分子量10w)水溶液;
[0066]
s3、将油相通过膜乳化器乳化进入转动的水相中,控制油相与水相的体积比为1/5,乳化压力0.04mpa,膜管长度为15μm,乳化时长为120min;磁子长度为5cm,乳化搅拌转速为350r/min。乳化完成后,开始固化,固化使用顶置搅拌器、10cm搅拌桨,另加入280ml水,搅拌转速为120r/min,固化时长24h,得到可注射pha微球。
[0067]
比较例2
[0068]
s1、制备油相:将0.8g的p34hb溶于20ml的二氯甲烷中,配制40mg/ml的pha/有机溶剂溶液,其中,p34hb的重均分子量为15万;
[0069]
s2、制备水相:配制质量分数为3%透明质酸(分子量7w)水溶液;
[0070]
s3、将油相通过膜乳化器乳化进入转动的水相中,控制油相与水相的体积比为1/5,乳化压力0.04mpa,膜管长度为15μm,乳化时长为120min;磁子长度为5cm,乳化搅拌转速为350r/min。乳化完成后,开始固化,固化使用顶置搅拌器、10cm搅拌桨,另加入280ml水,搅拌转速为120r/min,固化时长24h,得到可注射pha微球。
[0071]
测试例1
[0072]
由实施例1制得的可注射pha微球的电镜图可以看出,所得微球具有良好的分散性,相互之间没有粘连,且形态均一,为球形。
[0073]
接下来,用激光粒度仪进行粒径和span值的测试,图3为实施例1制得的可注射pha微球的粒径分布图。由图3可知,实施例1制得的微球平均粒径在33.7微米,表1是激光粒度仪测得的实施例1的span值(span值为粒径分布离散程度),检测结果显示span值为0.801,此结果表明,实施例1的可注射pha微球的粒径均一。
[0074]
表1
[0075][0076]
测试例2
[0077]
为了进一步检测可注射pha微球的蛋白吸附性,进行下述实验:
[0078]
(1)绘制蛋白生长曲线:用bsa配成3mg/ml的溶液,用分光光度计全波长扫描,检测bsa白蛋白的最大吸收波长为278.58nm。然后配置一系列浓度梯度的溶液0.5、1、1.5、3、4、5mg/ml,在最大吸收波长处检测各浓度的溶液的吸光值,绘制标准曲线,标准曲线如图4所示,图4的横坐标用x表示,纵坐标用y表示,x为bsa浓度,y为吸光值。
[0079]
(2)配置5mg/ml的bsa溶液,孵育可注射pha微球,孵育前,测得bsa溶液的吸光值为2.941,孵育5个小时后,离心取上清液,检测bsa溶液(上清溶液)的吸光值,计算具体的吸附浓度,结果为:孵育5小时后,上清液中bsa的浓度为4.79mg/ml,吸光值为2.892,孵育前后的浓度差表明了bsa蛋白被可注射pha微球吸附。
[0080]
(3)再计算每克微球吸附蛋白的值(数值越高,表示蛋白吸附性越好)。
[0081]
计算得到实施例1中可注射pha微球吸附蛋白的值为3600ug/g,同ct1电荷球(ct1电荷球是常用的细胞培养微载体,英文名为cytodex 1,ct1的主要成分是ge healthcare,deae-交联葡聚糖)的吸附值600ug/g比较,可见本实施例1提供的可注射pha微球能够很好地吸附在细胞表面,且吸附效果比传统的ct1效果要好,约是ct1球吸附能力的10倍。
[0082]
表2
[0083]
组别bsa吸附(μg/g)实施例13600实施例24000对比例11200对比例2900
[0084]
生物材料与生理环境相接触时,生物材料想要在体内不被排斥,没有严重的填充物相关副作用,需要很好的细胞粘附性,而细胞粘附性中一个很重要的点,在于细胞表面粘附有大量蛋白,而蛋白吸附是一个重要的细胞粘附推动力。ct1电荷球是常用的细胞培养微载体,数值越高,蛋白吸附性越好。经测试ct1电荷球吸附牛血清白蛋白的值600μg/g。由表2可知,实施例1制备的pha微球对牛血清白蛋白的吸附浓度为3600μg/g;实施例2制备的pha微球对牛血清白蛋白的吸附浓度为4000μg/g。经对比分析,本发明制备的pha微球具有优异的蛋白吸附性,吸附浓度是ct1电荷球的6-7倍,进而表明其具有优异的细胞粘附性,可以用于医疗、美容填充、植入医疗器械等。
[0085]
测试例3
[0086]
采用安东帕mcr302流变仪测定微球杨氏模量。首先将仪器设置到恒温振幅扫描模式,选择合适直径(25mm)的平板转子及加样台高度h(1mm),固定频率(1hz),然后设置剪切应变γ从0.01%至10%按对数变化。具体检测步骤为:
[0087]
将微球样本放置于加样台中心,转子下降至测量位置,刮样后开始测定,周围加盖
密封装置以减少测量过程中的水分损失,得到储能模量g'及损耗模量g"随剪切应变变化的曲线图。根据下列公式计算出杨氏模量e;e=2g(1 v),v=0.5
[0088]
表3
[0089]
组别杨氏模量(pa)实施例11108.4实施例21198.3对比例1100.3对比例290.4
[0090]
测试例4
[0091]
为模仿生理状态下体液循环对降解度的影响,根据蠕动泵原理,设计动态降解系统进行体外溶解实验。将5g微球置于200ml生理盐水中,37℃恒温水浴,用两个输液管分别作为滴入管和滴出管,调整滴入和流出速度,使之同时达到1000ml/h。分别于50、100、150、200、250、300、350、400天观测微球的降解情况,取混合液离心,如有沉淀物微球,则未降解完,如无沉淀物,则已降解完全,记录完全降解时间。实验表面使用交联透明质酸,pha微球降解时间得到了极大的增长。
[0092]
表4
[0093]
组别降解时间(天)实施例1300实施例2350对比例1200对比例2150
[0094]
尽管本发明已经参考示例性实施方案进行了描述,但应理解本发明不限于公开的示例性实施方案。在不背离本发明的范围或精神的情况下,可对本发明说明书的示例性实施方案做多种调整或变化。权利要求的范围应基于最宽的解释以涵盖所有修改和等同结构与功能。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
