1.本发明属于废水处理技术领域,尤其涉及吸收环氧氯丙烷的生产尾气产生的吸收液的处理。
背景技术:
2.环氧氯丙烷(c3h5clo,epichlorohydrin)是制造环氧树脂的重要原料,也是硝化甘油炸药、玻璃钢、电绝缘制品的主要原料。目前环氧氯丙烷的生产工艺有丙烯高温氯化法、醋酸丙烯酯法、丙烯醛法和甘油氯化法,然而上述工艺都伴随产生大量含盐、高cod废水,环境污染严重,已被列入高环境风险生产工艺目录。
3.近年来,研究人员提出以钛硅分子筛为催化剂、氯丙烯和双氧水为原料的直接氧化法,此方法原子利用率高、废水少,符合绿色化学发展方向,具有较好的工业化前景。但是采用此方法合成环氧氯丙烷的过程中会产生一定量的副产物氯甘油(c3h7clo2,3-氯丙二醇),该副产物主要于水相。
4.另一方面,由于环氧氯丙烷分子中含有环氧基团,化学性质极其活泼,具有燃爆危险性,因此被列入危险化学品名录。在以环氧氯乙烷为原料的生产过程中,为了避免尾气中残留的环氧氯丙烷发生泄漏、燃爆等危险性,常常将尾气通入碱性吸收液中,使其水解转化为氯甘油以消除其危害性。
5.氯甘油已被国际组织认定为高毒性污染物,多项研究表明氯甘油具有生殖、肾脏和神经毒性以及潜在的致癌和致突变作用,危害皮肤、眼、呼吸道,可致多种哺乳动物可逆性不育,在2011年国际癌症研究机构将氯甘油列为b类可能致癌物。因此含一定浓度氯甘油的废水属于危险废物,禁止直接排放。
6.然而,由于含氯有机物自身的毒性高会导致微生物失活,因而直接采用传统的活性污泥法废水处理方法对于含氯甘油废水的效果不佳。为此,现有技术采用吸附法或化学法与生化法组合工艺来处理。
7.吸附法的工作原理是利用特定吸附剂进行选择性吸附,以实现含氯有机物的回收和水的资源化利用。比如,专利文献1中公开了采用zn、fe、cu或者ag改性后颗粒状活性炭作吸附剂处理环氧氯丙烷生产废水的方法,然而吸附后需要加入二氯甲烷、丙酮等低沸点溶剂回收含氯有机物,溶剂损耗量大,运行成本高。
8.化学法的工作原理是利用化学方法将水中的氯甘油转化为其他物质。如专利文献2和专利文献3所述,采用碱性水溶液或固体强碱催化剂使废水中的含氯有机发生水解而脱氯,例如3-氯-1,2-丙二醇转化为甘油,或一氯丙二醇单甲醛转化为甘油单甲醛,实现废水的无毒无害化处理。然而,此方法实现可以含氯有机物的脱氯,但脱氯后的有机物仍残留在水中,因而后续仍需要其他水处理技术(如:萃取或生化法)才能符合排放标准。
9.因此,如何高效地去除吸收环氧氯丙烷的生产尾气产生的吸收液中的氯甘油是本领域亟待解决的技术问题。
10.专利文献
11.专利文献1:cn109456289a
12.专利文献2:cn106630083a
13.专利文献3:cn106630007a
技术实现要素:
14.针对现有的吸收环氧氯丙烷的生产尾气产生的吸收液中的氯甘油的去除方法所存在的问题,本发明人等经过了深入研究后发现,通过采用特定的阳极电极,将氯甘油转化为氯乙酸,后续利用氯乙酸在碱性水中溶解度低的特点采用结晶法析出,从而高效地进行氯甘油的去除,实现废弃资源的高效利用,并且不会引入需要进一步处理的其他成分,由此完成了本发明。
15.本发明涉及下述的技术方案
16.[1]、一种金基修饰电极,其通过含有如下步骤的制备方法制成:
[0017]
铜沉积步骤:在金片、金箔或表面镀金的导电基体上,在低于铜沉积电位的电位下进行恒电位沉积,沉积时间为1~50秒;
[0018]
第一修饰金属置换步骤:将经过了所述铜沉积步骤后的电极表面清洗后,放入含有第一修饰金属的离子的溶液中反应1~20分子,将电极表面的铜原子置换为第一修饰金属,前述第一修饰金属为选自钯、铂中的至少一种金属;
[0019]
第二修饰电极置换步骤:在经过了所述第一修饰金属置换步骤的电极上,进一步通过电化学沉积的方法,沉积上第二修饰金属,所述第二修饰金属为选自锡、锑、铟中的至少一种金属。
[0020]
[2]、根据[1]所述的金基修饰电极,其中,前述第二修饰金属置换步骤中,电化学沉积的时间为10~50秒。
[0021]
[3]、一种氯甘油的去除方法,其使用前述的[1]或[2]所述的金基修饰电极作为阳极,将氯甘油通过电解电化学方法氧化为氯乙酸。
[0022]
[4]、根据[3]所述的去除方法,其中,所述电化学方法中,采用不锈钢电极、钛电极、铂电极或石墨电极作为阴极。
[0023]
[5]、根据[3]或[4]所述的去除方法,其中,所述电化学方法中,采用恒电位或恒电流方式电解。
[0024]
[6]、根据[5]所述的去除方法,其中,采用恒电位方式电解,阳极电位为1.0~1.4v vs.sce。
[0025]
[7]、根据[3]~[6]的任一项所述的去除方法,其中,所述氧化的时间为1~4小时。
[0026]
[8]、根据[3]~[7]的任一项所述的去除方法,其还含有下述步骤:
[0027]
含有氯甘油的废碱液以间歇或流动方式加入电解槽中进行电解;
[0028]
根据电解液中氯甘油、氯乙酸的含量变化情况调节电解条件,判断转化程度;以及,
[0029]
采用冷却、浓缩或配位萃取结晶法使电解后的溶液中的氯乙酸盐析出。
[0030]
[9]、[3]~[8]的任一项所述的去除方法用于去除吸收环氧氯丙烷的生产尾气产生的吸收液中的氯甘油的用途。
附图说明
[0031]
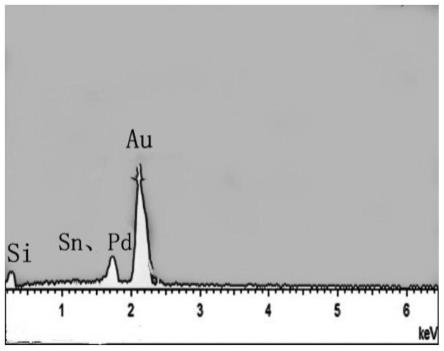
图1为实施例2中制备的sn-pd修饰镀金硅片电极的扫描电镜图。
[0032]
图2为实施例2中制备的sn-pd修饰镀金硅片电极的eds图。
具体实施方式
[0033]
[金基修饰电极]
[0034]
本发明的金基修饰电极为通过含有如下步骤的制备方法制成的电极。
[0035]
铜沉积步骤:在金片、金箔或表面镀金的导电基体上,在低于铜沉积电位的电位下进行恒电位沉积,沉积时间为1~50秒;
[0036]
第一修饰金属置换步骤:将经过了所述铜沉积步骤后的电极表面清洗后,放入含有第一修饰金属的离子的溶液中反应1~20分钟,将电极表面的铜原子置换为第一修饰金属,前述第一修饰金属为选自钯、铂中的至少一种金属;
[0037]
第二修饰电极置换步骤:在经过了所述第一修饰金属置换步骤的电极上,进一步通过电化学沉积的方法,沉积上第二修饰金属,前述第二修饰金属为选自锡、锑、铟中的至少一种金属。
[0038]
前述的导电集体是指金以外的其他金属或者半导体,如硅片。
[0039]
在使用金片、金箔或表面镀金的导电基体进行前述的铜沉积步骤之前,优选对其进行清洗。前述的清洗可以为例如,用通常的有机溶剂进行超声洗涤等方法,从而使得金原子晶面暴露。前述的有机溶剂可以列举出例如丙酮、乙醇等。
[0040]
例如,可以采取下述的设置:以饱和甘汞电极(sce)为参比电极、铂片电极为对电极,以上述基体电极为工作电极组成三电极体系,以浓度10-3
~1mol/l的cu
2
溶液(以h2so4调节溶液至弱酸性)为电解液溶液。
[0041]
前述的铜沉积步骤中,在低于铜沉积电位的电位下进行恒电位沉积,也即进行所谓的欠电位沉积。因此,在沉积铜之前,先确定铜的沉积电位。例如,10-3
mol/lcu
2
溶液的情况下,沉积电位为0.04v vs sce,然后控制工作电极恒定于该沉积电位,例如设置为0.03v vs sce,进行恒电位沉积。沉积时间根据电解液中cu
2
浓度及电极电位的,控制在1~50秒的范围。
[0042]
前述的第一修饰金属置换步骤中,将经过了铜沉积步骤后的电极表面清洗后,放入含有第一修饰金属的离子的溶液中反应1~20分钟,将电极表面的铜原子置换为第一修饰金属。前述第一修饰金属为选自钯、铂中的至少一种金属。
[0043]
前述的电极表面清洗采用惯常的电极清洗的方法,没有特别的限定。例如可以列举出用通常的有机溶剂进行超声洗涤等方法。前述的有机溶剂可以列举出例如丙酮、乙醇等。
[0044]
前述的含第一修饰金属的离子的溶液可以为例如pdcl2溶液、ptcl4溶液、等。前述的含第一修饰金属的离子的溶液用盐酸调节溶液至弱酸性(ph小于7)。
[0045]
前述的第一修饰金属置换步骤中,将电极有效面积部分浸入液面下,磁力搅拌下反应1~20分钟,使电极表面的铜原子被置换为第一修饰金属。
[0046]
前述的第二修饰电极置换步骤中,将经过了所述第一修饰金属置换步骤的电极作为工作电极,以含前述第二修饰金属离子的溶液(以h2so4调节溶液至弱酸性)为电解液进一
步通过电化学沉积的方法,沉积上第二修饰金属,前述第二修饰金属为选自锡、锑、铟中的至少一种金属。
[0047]
前述的第二修饰电极置换步骤中,可以采用例如以饱和甘汞电极为参比电极、铂片电极为对电极组成三电极体系。
[0048]
前述的含前述第二修饰金属离子的溶液可以为例如含sn
2
、sb
3
、或者、in
3
的溶液。
[0049]
前述的第二修饰电极置换步骤的沉积时间可以根据需要的第二修饰金属的沉积量以及第二修饰金属与第一修饰金属的比例来适宜调节,可以为例如10~50秒。
[0050]
[氯甘油的去除方法]
[0051]
本发明的氯甘油的去除方法中,使用前述的金基修饰电极作为阳极,将氯甘油通过电解电化学方法氧化为氯乙酸。
[0052]
本发明的氯甘油的去除方法中,氯甘油在作为阳极的金基修饰电极表面发生氧化,分子中两个羟基氧化为羧基同时发生c-c键断裂,生成氯乙酸和甲酸,甲酸随即分解为co2和h2o。化学方程式如式1所示。
[0053]
ch2cl-choh-ch2oh
→
ch2cl-cooh hcooh
ꢀꢀ
式1
[0054]
另外,氯甘油分子在阳极表面也有可能发生下述的式2~4所示的副反应。
[0055]
ch2cl-choh-ch2oh
→
ch2cl-choh-cooh(3-氯-2-羟基丙酸)
ꢀꢀ
式2
[0056]
ch2cl-choh-ch2oh
→
ch2cl-cho hcho(一氯乙醛)
ꢀꢀ
式3
[0057]
ch2cl-choh-ch2oh
→
ch2cl-choh-cho(3-氯-2-羟基丙醛)
ꢀꢀ
式4
[0058]
因此,在电化学氧化过程中,水中的氯甘油分子扩散到阳极表面,根据不同的氧化反应途径,两个羟基的氧化程度不同或者c-c键是否断裂的情况也不同,从而产生不同的氧化产物。
[0059]
通过采用前述的本发明的金基修饰电极作为阳极,能够减少前述的式2~式4所示的副反应的发生,高选择性地转化为氯乙酸。
[0060]
用于吸收环氧氯丙烷的生产尾气的吸收液通常为碱性水溶液,例如,氢氧化钠水溶液。吸收液中碱的存在不但有利于环氧氯丙烷的水解,还有利于降低电解时的溶液电阻。特别是,碱性介质有利于氯乙酸钠的析出,室温条件下,氯乙酸在水中的溶解度约为500g/100ml,氯乙酸钠的溶解度约为80g/100ml。因此,氧化生成的氯乙酸可以通过浓缩或冷却等方式析出,从而可以在降低水中含氯有机物含量的同时获得氯乙酸钠产品。
[0061]
另外,通过采用本发明的氯甘油的去除方法,处理过程中不外加试剂,避免了二次污染,可以得到高纯度的氯乙酸钠,从而降低废水处理成本。
[0062]
由于作为产物的氯乙酸具有足够的稳定性,因此,前述的氯甘油的去除方法中所使用的电解槽:无需用于分隔阴极室与阳极室的石棉、布或离子选择性透过膜等任何隔膜,可以采用圆柱形、方形等不同形状、不同体积的容器作为电解槽。
[0063]
前述的氯甘油的去除方法中,阴极可选用常见的不锈钢、钛、铂、石墨等材质制作的析氢电极或者能够使氧气发生2电子或4电子还原过程的氧气还原电极。电极形状和有效面积可根据需要而定,以片状电极为宜。阴极和阳极面积相当为宜。两电极相互平行为宜。两极之间距离尽量小为宜。
[0064]
前述的氯甘油的去除方法中,电解过程可以采用恒电位或恒电流方式,优选采用
恒电位方式。若采用恒电位电解方式,阳极电位可以控制在例如1.0~1.4v vs.sce的范围。若采用恒电流方式,则电流密度根据水中氯甘油浓度进行调节。电解时间为例如1~4小时。
[0065]
前述的氯甘油的去除方法中,可以采用间歇或流动方式将含有氯甘油的废水加入电解槽中。间歇方式是一次性注入后进行电解,电解完成后放出进行后续处理。流动方式是将待电解废水以一定流速不断泵入电解槽,并以一定流速流出。
[0066]
前述的氯甘油的去除方法中,还可以含有下述步骤:通过定期或不定期地从电解槽中取样,测定其中氯甘油和氯乙酸的含量变化情况来判断电化学转化程度。
[0067]
对于水中氯甘油和氯乙酸浓度的测定,可以根据现有技术进行测定,优选采用高效液相色谱法。进一步,优选选用对小分子有机糖、酸、醇表现良好的分离效果的色谱柱,例如美国伯乐公司的bio-rad aminex hpx-87h;以5mmol/l h2so4作为流动相,使待测物氯乙酸保持分子状态;选用示差折光检测器,可以实现同时检测氯甘油和氯乙酸。
[0068]
根据电解前溶液中氯甘油和氯乙酸浓度测定值,依据下列公式求算氯甘油转化率、氯乙酸收率及氯乙酸选择性。
[0069][0070][0071]
氯乙酸选择性=氯乙酸收率/氯甘油转化率
[0072]
对于电解后的溶液中的氯乙酸盐,可以采用冷却、浓缩或配位萃取结晶法使其析出。对于冷却、浓缩或配位萃取结晶法的具体步骤及条件,本领域技术人员能够根据需要适宜的设定,在此不再赘述。
[0073]
实施例
[0074]
实施例1制备sn-pd修饰金盘电极
[0075]
在电化学工作站(chi 660)上采用三电极体系进行:工作电极为盘状金电极(φ=3mm),使用前依次在丙酮和去离子水中超声,再用浓hno3溶液浸泡,最后用去离子水彻底冲洗并在氮气气流下干燥;对电极为铂片电极(15mm
×
15mm);以饱和甘汞电极为参比电极。具体电沉积步骤如下:
[0076]
以1mmol/l cuso4和0.1mol/l h2so4混合溶液为支持电解质溶液,从开路电位-0.15v(vs sce)起正向扫描电位,确定cu的沉积电位为0.04v;继而控制工作电极电位负于0.03v下进行欠电位沉积20s,取出电极后,依次使用0.1mol/l h2so4、去离子水以及少量乙醇清洗烘干,备用。
[0077]
将上述得到的电极浸入1mmol/l pdcl2与0.1mol/l hcl的混合溶液的液面以下,磁力搅拌1min,此时电极表面的cu原子被置换为pd原子。取出电极后,依次使用0.1mol/l hcl溶液、去离子水以及少量乙醇清洗,烘干备用。
[0078]
在含1mmol/l sn
2
的0.1mol/l h2so4溶液中,以上述修饰电极为工作电极在-0.277v电位下进行sn沉积,通过控制沉积时间在10~50s,从而调节电极上负载的sn和pd比例(pd:sn摩尔比在1:3至3:1之间),以上述方法清洗干燥后,即可得到下的表1所示的sn-pd修饰金盘电极。
[0079]
实施例2制备sn-pd修饰镀金硅片电极
[0080]
在电化学工作站(chi 660)上采用三电极体系进行:以镀金硅片电极(15mm
×
15mm)为工作电极,对电极为铂片电极(15mm
×
15mm);以饱和甘汞电极为参比电极。具体电沉积步骤如下:
[0081]
以5mmol/l cuso4溶液(硫酸调节ph 1.0左右)为支持电解质溶液,控制工作电位为0.03v进行欠电位沉积cu 20s,取出电极后,依次使用0.1mol/l h2so4溶液、去离子水以及少量乙醇清洗烘干,备用。
[0082]
将上述得到的电极浸入5mmol/l pdcl2(盐酸调节ph 1.0左右)液面以下,磁力搅拌1min,此时电极表面的cu原子被置换为pd原子。取出电极后,依次使用0.1mol/l盐酸溶液、去离子水以及少量乙醇清洗,烘干备用。
[0083]
将sno溶解于0.1mol/l h2so4溶液,配制为5mmol/l sn
2
溶液(ph 1.0左右)作为电解液,以上述修饰电极为工作电极在-0.30v电位下沉积30s,以上述方法清洗干燥后,即可得到sn-pd修饰镀金硅片电极(其中sn:pd比例为1:1)。
[0084]
图1和图2分别示出了制备得到的sn-pd修饰镀金硅片电极的扫描电镜图及eds图。图1及图2表明,sn-pd修饰镀金硅片电极的镀金硅基底上负载有金属sn和pd。
[0085]
实施例3~7电化学法氧化氯甘油试验
[0086]
在50ml小烧杯中加入20ml含有0.1mol/l氯甘油的碱性溶液(naoh浓度为1mol/l),以前述实施例1制备的不同pd/sn比例的sn-pd修饰金盘电极为阳极,铂片电极为阴极。以饱和甘汞电极为参比电极,控制阳极电位为1.35v进行恒电位电解4h。电解过程中持续搅拌电解液。完成电解后电解槽中取样,采用高效液相色谱法hplc测定其中氯甘油和氯乙酸的含量,计算氯甘油转化率、氯乙酸收率及氯乙酸选择性如表1所示。
[0087]
表1不同pd/sn比例的sn-pd修饰金盘电极电化学法氧化氯甘油效果
[0088][0089]
实施例8~10
[0090]
在50ml小烧杯中加入20ml含有0.1mol/l氯甘油的碱性溶液(naoh浓度为1mol/l),以pd/sn比例为1:1的sn-pd修饰金盘电极为阳极,铂片电极为阴极。以饱和甘汞电极为参比电极,控制阳极电位为1.35v进行恒电位分别电解1h、2h和4h。电解过程中持续搅拌电解液。完成电解后电解槽中取样,采用高效液相色谱法hplc测定其中氯甘油和氯乙酸的含量,计算氯甘油转化率、氯乙酸收率及氯乙酸选择性如表2所示。
[0091]
表2不同电解时间的电化学法氧化氯甘油效果
[0092][0093]
实施例11~13不同碱度下的电化学法氧化氯甘油效果试验
[0094]
在50ml小烧杯中加入20ml含有0.1mol/l氯甘油的碱性溶液,其中naoh质量浓度分别0.05%、0.1%、0.5%,以pd/sn比例为1:1的sn-pd修饰金盘电极为阳极,铂片电极为阴极。以饱和甘汞电极为参比电极,控制阳极电位为1.35v进行恒电位分别电解2h。电解过程中持续搅拌电解液。完成电解后电解槽中取样,采用高效液相色谱法hplc测定其中氯甘油和氯乙酸的含量,计算氯甘油转化率、氯乙酸收率及氯乙酸选择性如表3所示。
[0095]
表3不同碱度下的电化学法氧化氯甘油效果
[0096][0097]
实施例14萃取结晶法析出氯乙酸钠
[0098]
将三辛胺溶解于正己烷,配成100~200g/l溶液,将含有0.3~0.5mol/l氯乙酸钠的电解液与有机溶液的体积比1:1在30~50℃下混合搅拌0.5~1.0h,静置分层;在分离后的水相中,继续加入1-3倍体积的正己烷,在30~50℃下搅拌约0.5-1.0h,静置分层取水相;多次萃取后,蒸干水相,得到的固体即为氯乙酸钠。氯乙酸钠结晶率可达98.85%,纯度达到97.1%,母液中残留含氯有机物的量不超过9.72mg/l。
[0099]
以上详细描述了本发明的优选实施方式,但是,本发明并不限于此。在本发明的技术构思范围内,可以对本发明的技术方案进行多种简单变型,包括各个技术特征以任何其它的合适方式进行组合,这些简单变型和组合同样应当视为本发明所公开的内容,均属于本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。