1.本发明涉及精细化学品合成领域,尤其涉及一种乙醛偶姻缩合非均相催化剂及其制备方法和应用。
背景技术:
2.乙偶姻,又名3-羟基-2-丁酮、甲基乙酰甲醇、醋嗡,具有令人愉快的奶油香气,被广泛应用于葡萄酒、奶油、酸奶、蜂蜜、草莓等型香精的配制,是我国批准使用的香料产品(gb 2760-2014)。此外,乙偶姻还可用于修饰青霉素、氨苄青霉素等抗生物素药物,以提高药效、减轻药物副作用。
3.传统乙偶姻制备方法有:
①
加氢还原/氧化合成法,以2,3-丁二酮或2,3-丁二醇为原料,部分加氢还原或部分氧化制备乙偶姻,但原料成本较高,收率和产品质量都不理想。
②
生物发酵法,使用山梨糖菌在2,3-丁二醇中发酵,或使用曲霉菌、青霉菌等在甘蔗汁中发酵获得乙偶姻,但方法收率低、产物回收困难、投资大、规模不易扩大。
③
乙醛催化缩合法,利用乙醛的自生压力,在催化剂作用下进行偶姻缩合反应一步偶联制得乙偶姻,原子经济性好,符合绿色化学需求,原料成本低,具有良好的应用前景。
4.早期偶姻缩合反应使用的催化剂为氢氰酸盐,价格低廉且催化反应产物纯度高,但由于其剧毒性,对人体和环境都存在较大风险,不适合于食用香料的工业合成。自breslow(j. am.chem.soc.80,3719(1959))提出将噻唑鎓用作乙醛偶姻缩合反应催化剂后,在此方面已有较多应用(cn 1562934a,cn 107188793等)。但是,残留在产物中的噻唑鎓盐难以去除,严重影响乙偶姻产品质量,同时催化剂无法回收利用,造成生产成本上升。
5.为降低产物纯化难度,同时实现催化剂的循环使用,有研究基于不同载体的负载制备非均相催化剂,实现乙醛偶姻缩合噻唑鎓催化剂的固定化(如专利wo2015112096a1采用氯甲基化聚苯乙烯树脂作为载体,专利cn112500366a采用石墨烯作为载体),但这些方法普遍存在噻唑鎓活性中心与载体之间通过刚性键连接,造成催化活性低的问题;并且,负载过程需要强酸或强碱的参与,极易发生噻唑环的开环反应,同样会导致最终获得的非均相催化剂催化活性过低;此外,催化剂合成步骤繁琐,制备成本高,难以实现大规模工业运用。
技术实现要素:
6.为了解决上述技术问题,本发明提供了一种乙醛偶姻缩合非均相催化剂及其制备方法和应用。该非均相催化剂通过柔性共价键连接催化活性中心和载体,且制备过程无需加入强酸强碱,因而具有较高的催化活性;此外还具有制备方法简单、成本低的优点,有利于实现大规模工业运用。
7.本发明的具体技术方案为:第一方面,本发明提供了一种乙醛偶姻缩合非均相催化剂,结构通式如下:
其中,r1和r2分别独立地选自氢、1-8个碳原子的烷基、2-8个碳原子的烯基、6-8个碳原子的芳基、1-8个碳原子的烷氧基、羟基取代的烷基、羟基取代的烯基、羟基取代的芳基或羟基取代的烷氧基。
8.本发明的非均相催化剂以噻唑鎓为活性中心,通过将催化活性中心与载体相连,实现噻唑鎓催化剂的固定化。对于噻唑鎓催化剂而言,其催化活性与卡宾碳周围的空间位阻和电子云密度密切相关,在将催化活性中心与载体连接后,载体易影响卡宾碳活性位点的电子云密度,并对其造成较大的空间位阻,进而对催化活性产生不利影响。而本发明的非均相催化剂以柔性饱和脂肪烃链连接噻唑鎓活性中心与载体,能够在维持活性中心电荷分布的同时,减小连接部位对卡宾碳造成的空间位阻,因而具有较高的催化活性。
9.并且,本发明的非均相催化剂采用一步自由基聚合反应即可合成,制备过程简单、成本低,能够实现规模化生产,更重要的是,聚合反应过程中条件温和,无需加入强酸强碱,因而能够减少噻唑环的开环反应,使非均相催化剂具有较高的催化活性。
10.作为优选,r1和r2均为甲基。
11.当r1和r2为甲基时,在非均相催化剂的制备过程中,这两个非极性的甲基能够与二乙烯基苯产生较强的疏水相互作用,使得对位的卡宾碳活性位点定向、均匀地分布在非均相催化剂的孔道内,从而在较大程度上发挥噻唑鎓活性中心的催化能力,提高非均相催化剂的催化活性。
12.作为优选,x-为f-、cl-、br-、i-、bf
4-、clo
4-或no
3-。
13.第二方面,本发明提供了一种所述非均相催化剂的制备方法,包括以下步骤:将含烯基的催化活性物种、二乙烯基苯、致孔溶剂和聚合引发剂混合,进行聚合反应后,分离产物,获得乙醛偶姻缩合非均相催化剂;所述含烯基的催化活性物种的结构通式如下:
14.含烯基的催化活性物种结构通式中的r1、r2和x即非均相催化剂中的r1、r2和x。
15.作为优选,所述含烯基的催化活性物种、二乙烯基苯、致孔溶剂和聚合引发剂质量比为1:(1-50):(2-200):(0.001-0.5)。
16.在本发明催化剂的制备过程中,二乙烯基苯、致孔剂溶液和聚合引发剂的用量均会影响最终获得的催化剂性能,具体而言: 1
○
二乙烯基苯:在含烯基的催化活性物种中,噻唑环具有较大的空间位阻,当二乙烯基苯的用量过少时,难以得到具有一定聚合度的固体产物,不利于催化剂在乙醛偶姻缩合反应中的使用和回收;而当二乙烯基苯的用量过大
时,会造成制得的催化剂中催化位点密度过低,影响催化活性。
[0017]2○
致孔溶剂:致孔溶剂能够使非均相催化剂中形成孔道,提高催化剂的比表面积,从而提高催化活性。但当致孔溶剂的用量过大时,为了获得具有一定聚合度的固体产物,需要相应地延长聚合反应时间,这会造成产物形态难以控制,进而导致非均相催化剂的催化活性不理想。
[0018]3○
聚合引发剂:当引发剂用量过小时,需要相应地延长聚合反应时间,造成制得的非均相催化剂形态难以控制,影响其催化活性;当引发剂用量过大时,会造成聚合反应速度过快,制得的催化剂聚合度过低,不利于催化剂在乙醛偶姻缩合反应中的使用和回收。
[0019]
本发明综合考虑上述因素,将烯基的催化活性物种、二乙烯基苯、致孔溶剂和聚合引发剂质量比控制在1:(1-50):(2-200):(0.001-0.5)范围内,能使制得的非均相催化剂兼具较高的聚合度和较好的形态,因而具有较高的催化活性,且便于在乙醛偶姻缩合反应中的使用和回收。
[0020]
作为优选,对于x为卤素的方案,所述含烯基的催化活性物种的制备方法包括以下步骤:以噻唑双取代衍生物和ch2=chch2x为原料,进行n-烷基化反应后,分离产物,获得含烯基的催化活性物种;所述噻唑双取代衍生物的结构通式如下:
[0021]
噻唑双取代衍生物结构通式中的r1和r2即非均相催化剂中的r1和r2;ch2=chch2x 中的x即非均相催化剂中的x。
[0022]
作为优选,对于x为非卤素的方案,所述含烯基的催化活性物种的制备方法包括以下步骤:以噻唑双取代衍生物和ch2=chch2y为原料,进行n-烷基化反应后,分离产物,获得n-烷基化反应产物;将n-烷基化反应产物与x-进行离子交换,分离产物,获得含烯基的催化活性物种;所述ch2=chch2y中,y为卤素;所述噻唑双取代衍生物的结构通式如下:
[0023]
噻唑双取代衍生物结构通式中的r1和r2即非均相催化剂中的r1和r2;“将n-烷基化反应产物与x-进行离子交换”中的x-即非均相催化剂中的x-。
[0024]
作为优选,所述聚合反应的温度为60-240℃,时间为2-24h。
[0025]
作为优选,所述致孔溶剂包括四氢呋喃、乙酸乙酯、丙酮、n,n-二甲基甲酰胺、n-甲基吡咯烷酮和1,2-二氯乙烷中的一种或多种。
[0026]
第三方面,本发明提供了所述非均相催化剂在乙醛偶姻缩合反应制备乙偶姻中的应用。
[0027]
作为优选,所述应用包括以下步骤:将所述非均相催化剂、碱性助剂和乙醛混合构成 ph为8-10的反应体系,在60-150℃下进行偶姻缩合反应,分离产物,制得乙偶姻。
[0028]
对于本发明的非均相催化剂而言,在催化乙醛偶姻缩合反应的过程中,催化剂卡
宾碳位点的去质子化以及噻唑环的开环分解均受反应条件的影响,反应体系的ph和反应温度过高和过低均会造成催化剂的催化活性过低,具体而言:当反应体系的ph过低或温度过低时,催化剂的卡宾碳位点难以去质子活化,导致活化活性过低;而当ph过高或温度过高时,会加速催化剂中噻唑环的开环分解,不仅会造成催化剂的偶姻缩合反应催化活性和选择性下降,还会产生较多难以去除的含硫杂质,造成制得的乙偶姻中存在异味,同时造成反应装置内出现结焦,不利于乙偶姻的分离纯化。
[0029]
进一步地,所述非均相催化剂与乙醛的质量比为1:(1-100)。
[0030]
进一步地,所述碱性助剂包括氢氧化钠、氢氧化钾、甲醇钠、氢化钠、碳酸钠、碳酸氢钠、碳酸钾、碳酸氢钾、甲胺、二乙胺和三乙胺中的一种或多种。
[0031]
进一步地,所述偶姻缩合反应的时间为0.5-6.0h。
[0032]
与现有技术相比,本发明具有以下优点:(1)本发明的非均相催化剂通过柔性共价键连接催化活性中心和载体,且制备过程无需强酸强碱参与,具有催化活性高、制备方法简单、生产成本低的优点;(2)本发明在非均相催化剂的制备过程中,通过控制二乙烯基苯、致孔溶剂和聚合引发剂的用量,在赋予非均相催化剂较高的催化活性的同时,有利于其在乙醛偶姻缩合反应中的使用和回收。
附图说明
[0033]
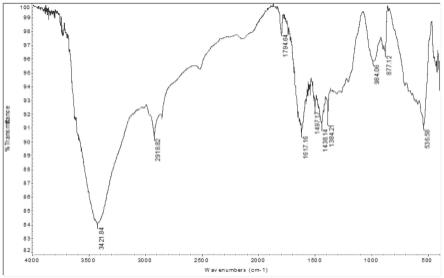
图1为制备例1中制得的非均相催化剂的红外光谱图;图2为备例1中制得的非均相催化剂的氮气吸附-脱附等温线图。
具体实施方式
[0034]
下面结合实施例对本发明作进一步的描述。
[0035]
总实施例一种乙醛偶姻缩合非均相催化剂,结构通式如下:其中,r1和r2分别独立地选自氢、1-8个碳原子的烷基、2-8个碳原子的烯基、6-8个碳原子的芳基、1-8个碳原子的烷氧基、羟基取代的烷基、羟基取代的烯基、羟基取代的芳基或羟基取代的烷氧基;x-为f-、cl-、br-、i-、bf
4-、clo
4-或no
3-。
[0036]
作为一种优选的实施方式,r1和r2均为甲基。
[0037]
当x-为f-、cl-、br-或i-时,通过以下步骤合成上述非均相催化剂:(1)以噻唑双取代衍生物和ch2=chch2x为原料,进行反应后,分离产物,获得含烯基的催化活性物种;所述噻唑双取代衍生物的结构通式如下:
所述含烯基的催化活性物种的结构通式如下:(2)将含烯基的催化活性物种、二乙烯基苯、致孔溶剂和聚合引发剂以 1:(1-50):(2-200):(0.001-0.5)的质量比混合,所述致孔溶剂包括四氢呋喃、乙酸乙酯、丙酮、n,n
‑ꢀ
二甲基甲酰胺、n-甲基吡咯烷酮和1,2-二氯乙烷中的一种或多种,在60-240℃下进行聚合反应2-48h后,分离产物,获得乙醛偶姻缩合非均相催化剂。
[0038]
当x-为bf
4-、clo
4-或no
3-时,通过以下步骤合成上述非均相催化剂:(1)以噻唑双取代衍生物和ch2=chch2y为原料,进行n-烷基化反应后,分离产物,获得 n-烷基化反应产物;将n-烷基化反应产物与x-进行离子交换,分离产物,获得含烯基的催化活性物种;所述ch2=chch2y中,y为卤素;所述噻唑双取代衍生物的结构通式如下:所述含烯基的催化活性物种的结构通式如下:(2)将含烯基的催化活性物种、二乙烯基苯、致孔溶剂和聚合引发剂以 1:(1-50):(2-200):(0.001-0.5)的质量比混合,所述致孔溶剂包括四氢呋喃、乙酸乙酯、丙酮、n,n
‑ꢀ
二甲基甲酰胺、n-甲基吡咯烷酮和1,2-二氯乙烷中的一种或多种,在60-240℃下进行聚合反应2-48h后,分离产物,获得乙醛偶姻缩合非均相催化剂。
[0039]
采用上述非均相催化剂,进行乙醛偶姻缩合反应制备乙偶姻,具体过程如下:将非均相催化剂、碱性助剂和乙醛混合构成ph为8-10的反应体系,所述非均相催化剂与乙醛的质量比为1:(1-100),所述碱性助剂包括氢氧化钠、氢氧化钾、甲醇钠、氢化钠、碳酸钠、碳酸氢钠、碳酸钾、碳酸氢钾、甲胺、二乙胺和三乙胺中的一种或多种,在60-150
℃下进行偶姻缩合反应0.5-6.0h,分离产物,制得乙偶姻。
[0040]
制备例1一种乙醛偶姻缩合非均相催化剂,结构式如下:其中,r1为甲基,r2为2-羟乙基,x-为br-。
[0041]
通过以下步骤合成上述非均相催化剂:(1)将5-(2-羟乙基)-4-甲基噻唑1g溶于1-溴丙烯5ml后,加热至80℃回流反应8h,冷却至室温后,旋转蒸发除去多余的1-溴丙烯,得到n-烯丙基-5-(2-羟乙基)-4-甲基噻唑溴盐;(2)将n-烯丙基-5-(2-羟乙基)-4-甲基噻唑溴盐1g、二乙烯基苯2g和偶氮二异丁腈100mg 加入到四氢呋喃15g中,混合搅拌后,加热至90℃,反应24h。过滤分离出固体产物,将固体产物使用四氢呋喃洗涤并烘干后,得到米黄色聚合物,即乙醛偶姻缩合非均相催化剂。
[0042]
对制得的浅黄色聚合物进行红外光谱测试,结果见图1。在图1中,2919cm-1
和1497 cm-1
处出现的强吸收峰表示有亚甲基存在,877cm-1
处出现较强的特征峰表示苯环是对位双取代,1384cm-1
处出现尖锐的峰表示碳氮键、碳硫键的存在,1617cm-1
处出现的强吸收峰表示季铵盐的结构存在,984cm-1
处出现的弱峰表示噻唑杂环上的碳氢键的存在。由此可证明催化剂成功负载。
[0043]
对制得的浅黄色聚合物进行77k氮气吸附和解吸试验,获得的氮气吸附-解吸等温线见图2,根据bet模型计算出所得非均相催化剂的比表面积为513m2/g。
[0044]
制备例2一种乙醛偶姻缩合非均相催化剂,结构式如下:其中,r1为甲基,r2为苄基,x-为br-。
[0045]
通过以下步骤合成上述非均相催化剂:(1)将5-苄基-4-甲基噻唑1g溶于1-溴丙烯5ml后,加热至80℃回流反应8h,冷却至室温后,旋转蒸发除去多余的1-溴丙烯,得到n-烯丙基-5-苄基-4-甲基噻唑溴盐;(2)将n-烯丙基-5-苄基-4-甲基噻唑溴盐1g、二乙烯基苯2g和偶氮二异丁腈100mg加入到四氢呋喃15g中,混合搅拌后,加热至90℃,反应24h。过滤分离出固体产物,将固体产
物使用四氢呋喃洗涤并烘干后,得到浅黄色聚合物,即乙醛偶姻缩合非均相催化剂,通过红外光谱测试证明催化剂成功负载。
[0046]
对制得的浅黄色聚合物进行77k氮气吸附和解吸试验,测得本制备例制得的非均相催化剂的比表面积为485m2/g。
[0047]
制备例3一种乙醛偶姻缩合非均相催化剂,结构式如下:其中,r1为甲基,r2为甲基,x-为br-。
[0048]
通过以下步骤合成上述非均相催化剂:(1)将4,5-二甲基噻唑1g溶于1-溴丙烯5ml后,加热至80℃回流反应8h,冷却至室温后,旋转蒸发除去多余的1-溴丙烯,得到n-烯丙基-4,5-二甲基噻唑溴盐;(2)将n-烯丙基-4,5-二甲基噻唑溴盐1g、二乙烯基苯2g和偶氮二异丁腈100mg加入到四氢呋喃15g中,混合搅拌后,加热至90℃,反应24h。过滤分离出固体产物,将固体产物使用四氢呋喃洗涤并烘干后,得到浅黄色聚合物,即乙醛偶姻缩合非均相催化剂,通过红外光谱测试证明催化剂成功负载。
[0049]
对制得的浅黄色聚合物进行77k氮气吸附和解吸试验,测得本制备例制得的非均相催化剂的比表面积为550m2/g。
[0050]
制备例4一种乙醛偶姻缩合非均相催化剂,结构式如下:其中,r1为甲基,r2为甲基,x-为br-。
[0051]
通过以下步骤合成上述非均相催化剂:(1)将4,5-二甲基噻唑1g溶于1-溴丙烯5ml后,加热至80℃回流反应8h,冷却至室温后,旋转蒸发除去多余的1-溴丙烯,得到n-烯丙基-4,5-二甲基噻唑溴盐;(2)将n-烯丙基-4,5-二甲基噻唑溴盐1g、二乙烯基苯1g和偶氮二异丁腈1mg加入到四氢呋喃2g中,混合搅拌后,加热至100℃,反应36h后出现固体产物。过滤分离出固体产物,将固体产物使用四氢呋喃洗涤并烘干后,得到浅黄色聚合物,即乙醛偶姻缩合非均相催化剂,通过红外光谱测试证明催化剂成功负载。
[0052]
对制得的浅黄色聚合物进行77k氮气吸附和解吸试验,测得本制备例制得的非均
相催化剂的比表面积为176m2/g。
[0053]
制备例5一种乙醛偶姻缩合非均相催化剂,结构式如下:其中,r1为甲基,r2为甲基,x为br。
[0054]
通过以下步骤合成上述非均相催化剂:(1)将4,5-二甲基噻唑1g溶于1-溴丙烯5ml后,加热至80℃回流反应8h,冷却至室温后,旋转蒸发除去多余的1-溴丙烯,得到n-烯丙基-4,5-二甲基噻唑溴盐;(2)将n-烯丙基-4,5-二甲基噻唑溴盐1g、二乙烯基苯50g和偶氮二异丁腈500mg加入到 n,n-二甲基甲酰胺200g中,混合搅拌后,加热至90℃,反应24h后出现固体产物。过滤分离出固体产物,将固体产物使用n,n-二甲基甲酰胺洗涤并烘干后,得到米白色聚合物,即乙醛偶姻缩合非均相催化剂,通过红外光谱测试证明催化剂成功负载。
[0055]
对制得的米白色聚合物进行77k氮气吸附和解吸试验,测得本制备例制得的非均相催化剂的比表面积为724m2/g。
[0056]
制备例6一种乙醛偶姻缩合非均相催化剂,结构式如下:其中,r1为甲基,r2为甲基,x-为bf
4-。
[0057]
通过以下步骤合成上述非均相催化剂:(1)将4,5-二甲基噻唑1g溶于1-溴丙烯5ml后,加热至80℃回流反应8h,冷却至室温后,旋转蒸发除去多余的1-溴丙烯,得到n-烯丙基-4,5-二甲基噻唑溴盐;(2)取n-烯丙基-4,5-二甲基噻唑溴盐1g溶于甲醇5ml后,加入与n-烯丙基-4,5-二甲基噻唑溴盐等摩尔量的四氟硼酸钠的饱和水溶液,室温下搅拌24h,所得产物旋转蒸发去除溶剂后水洗、干燥,得到n-烯丙基-4,5-二甲基噻唑四氟硼酸盐;(3)将n-烯丙基-4,5-二甲基噻唑四氟硼酸盐1g、二乙烯基苯2g和偶氮二异丁腈100mg加入到n,n-二甲基甲酰胺15g中,混合搅拌后,加热至90℃,反应24h后过滤分离出固体产物,将固体产物使用n,n-二甲基甲酰胺洗涤并烘干后,得到浅黄色聚合物,即乙醛偶姻缩合非均相催化剂,通过红外光谱测试证明催化剂成功负载。
[0058]
对制得的浅黄色聚合物进行77k氮气吸附和解吸试验,测得本制备例制得的非均
相催化剂的比表面积为724m2/g。
[0059]
制备对比例1-2按照制备例4中的步骤,合成制备对比例1和2的非均相催化剂。与制备例4的区别仅在于,按照表1,改变步骤(2)中二乙烯基苯和偶氮二异丁腈的用量,并记录出现固体产物的时间 (即反应时间)。
[0060]
表1
ꢀꢀ
二乙烯基苯/g偶氮二异丁腈/mg反应时间/h制备对比例10.51》72制备对比例210.248结果分析:在制备对比例1中,聚合反应72h后仍未形成固体产物,说明二乙烯基苯的用量过少会造成非均相催化剂难以制成。推测原因在于:在含烯基的催化活性物种(步骤(1)制得的n-烯丙基-4,5-二甲基噻唑溴盐)中,噻唑环具有较大的空间位阻,因此,当二乙烯基苯的用量过少时,难以得到具有一定聚合度的固体产物。
[0061]
制备对比例3-4按照制备例5中的步骤,合成制备对比例3和4的非均相催化剂。与制备例5的区别仅在于,按照表2,改变步骤(2)中二乙烯基苯和偶氮二异丁腈的用量,并记录出现固体产物的时间 (即反应时间)。
[0062]
表2
ꢀꢀ
偶氮二异丁腈/gn,n-二甲基甲酰胺/g反应时间/h制备对比例31.0200》24制备对比例40.550048结果分析:在制备例5中,聚合反应24h后形成了固体产物。相较于制备例5而言,制备对比例3增大了偶氮二异丁腈的用量,但反应24h后未形成固体产物,说明引发剂用量过大会造成非均相催化剂难以制成。推测原因在于:当引发剂用量过大时,会造成聚合反应速度过快,制得的催化剂聚合度过低,难以形成非均相催化剂。
[0063]
应用例1分别采用制备例1-6和制备对比例2、4获得的非均相催化剂,进行乙醛偶姻缩合反应制备乙偶姻,具体过程如下:将乙醛20g和非均相催化剂1g加入到50ml耐压反应釜中,加入碳酸氢钠调节ph至8,开启搅拌并升温至100℃,此时反应釜内压力达到1.5mpa,在100℃下反应5h。反应结束后,待温度降至室温,得到反应液18g。对反应液进行过滤,滤出固体物质,获得的液体即为乙偶姻产品。
[0064]
对乙偶姻产品进行成分分析,获得其中乙偶姻的含量后,计算乙醛转化率、乙偶姻选择性和乙偶姻总收率见表3。
[0065]
表3 非均相催化剂乙醛转化率/%乙偶姻选择性/%乙偶姻总收率/%制备例160.188.653.2制备例265.790.159.2制备例372.391.466.1
制备例470.480.957.0制备例556.189.750.3制备例671.291.064.8制备对比例243.855.324.2制备对比例439.250.519.8结果分析:(1)制备例1-3的非均相催化剂中,r1、r2存在差异,其中,制备例1的r1和r2分别为甲基和2-羟乙基,制备例2分别为甲基和苄基,制备例3均为甲基。从结果来看,相较于制备例1和2而言,当采用制备例3的非均相催化剂进行乙醛偶姻缩合反应时,乙偶姻总收率较高,说明制备例3的非均相催化剂具有较高的催化活性。推测原因在于:制备例3的非均相催化剂在制备过程中,噻唑环上4号位和5号位的两个甲基能够与二乙烯基苯产生较强的疏水相互作用,使得对位的卡宾碳活性位点定向、均匀地分布在非均相催化剂的孔道内,从而在较大程度上发挥噻唑鎓活性中心的催化能力,使最终制得的非均相催化剂具有较高的催化活性;此外,制备例2中的苄基存在柔性,难以有效地通过其与载体的疏水作用控制噻唑环的方向,因而对非均相催化剂催化活性的作用不如制备例3中的甲基。
[0066]
(2)在制备例3和制备对比例2中,偶氮二异丁腈的用量分别为1mg和0.5mg,其他原料和步骤均相同。从结果来看,制备对比例2虽然通过延长反应时间获得了非均相催化剂,但其催化活性明显低于制备例3。推测原因在于:当引发剂用量过小时,为了获得具有一定聚合度的固体产物,需要相应地延长聚合反应时间,这会造成制得的非均相催化剂形态难以控制,影响其催化活性。
[0067]
(3)在制备例4和制备对比例4中,n,n-二甲基甲酰胺的用量分别为200g和250g,其他原料和步骤均相同。从结果来看,制备对比例4虽然通过延长反应时间获得了非均相催化剂,但其催化活性明显低于制备例4。推测原因在于:当致孔溶剂的用量过大时,为了获得具有一定聚合度的固体产物,需要相应地延长聚合反应时间,这会造成产物形态难以控制,进而导致非均相催化剂的催化活性不理想。
[0068]
应用例2在应用例1中,使用制备例1获得的非均相催化剂进行乙醛偶姻缩合反应后,回收非均相催化剂,再次用于催化乙醛偶姻缩合反应制备乙偶姻,具体过程如下:在应用例1中,当使用制备例3获得的非均相催化剂进行乙醛偶姻缩合反应时,将步骤(2) 中滤出的固体物质使用乙醇洗涤并烘干,获得回收的非均相催化剂,与乙醛20g加入到50ml 耐压反应釜中,加入氢氧化钾调节ph至8,开启搅拌并升温至100℃,此时反应釜内压力达到1.5mpa,在100℃下反应5h后,反应釜内的压力下降至0mpa。反应结束后,待温度降至室温,得到反应液18g,滤出固体物质后,获得的液体即为乙偶姻产品。
[0069]
经检测,本应用例中,乙醛转化率为70.3%,乙偶姻选择性为90.7%,乙偶姻总收率为63.8%。
[0070]
应用例3回收应用例2中使用后的非均相催化剂,再次用于催化乙醛偶姻缩合反应制备乙偶姻,具体过程如下:将应用例2中滤出的固体物质使用乙醇洗涤并烘干后,与乙醛20g加入到50ml耐压
反应釜中,加入氢氧化钾调节ph至8,开启搅拌并升温至120℃,此时反应釜内压力达到1.5mpa,在120℃下反应5h后,反应釜内的压力下降至0mpa。反应结束后,待温度降至室温,得到反应液18g,滤出固体物质后,获得的液体即为乙偶姻产品。
[0071]
经检测,本应用例中,乙醛转化率为68.8%,乙偶姻选择性为90.4%,乙偶姻总收率为62.2%。
[0072]
结果分析:分析应用例2和3的数据可知,本发明的非均相催化剂可回收利用,在多次使用后仍能维持较高的催化活性和选择性。
[0073]
应用例4采用制备例3获得的非均相催化剂,进行乙醛偶姻缩合反应制备乙偶姻,具体过程如下:将乙醛20g和非均相催化剂1g加入到50ml耐压反应釜中,加入氢氧化钠调节ph至10,开启搅拌并升温至60℃,此时反应釜内压力达到1.0mpa,在60℃下反应6h后,反应釜内的压力下降至0mpa。反应结束后,待温度降至室温,得到反应液19g,滤出固体物质,获得的液体即为乙偶姻产品。
[0074]
经检测,本应用例中,乙醛转化率为69.6%,乙偶姻选择性为87.1%,乙偶姻总收率为60.6%。
[0075]
应用例5采用制备例3获得的非均相催化剂,进行乙醛偶姻缩合反应制备乙偶姻,具体过程如下:将乙醛20g和非均相催化剂1g加入到50ml耐压反应釜中,加入氢氧化钠调节ph至8,开启搅拌并升温至150℃,此时反应釜内压力达到1.5mpa,在150℃下反应3h后,反应釜内的压力下降至0mpa。反应结束后,待温度降至室温,得到反应液18g,滤出固体物质后,获得的液体即为乙偶姻产品。
[0076]
经检测,本应用例中,乙醛转化率为74.3%,乙偶姻选择性为66.5%,乙偶姻总收率为49.4%。
[0077]
应用对比例1本应用对比例中,按照应用例4中的步骤,进行乙醛偶姻缩合反应制备乙偶姻。与应用例4 的区别仅在于,将ph由10换成12。
[0078]
经检测,本应用对比例中,乙醛转化率为72.2%,乙偶姻选择性为39.5%,乙偶姻总收率为28.5%。
[0079]
结果分析:相较于应用例4而言,应用对比例1中乙偶姻产物的选择性和收率明显较低,推测原因在于:当反应体系的ph过高时,易造成非均相催化剂中的噻唑环开环分解,失去偶姻缩合反应催化活性,进而影响乙醛偶姻缩合反应的速率与选择性,同时,噻唑环开环分解还会产生难以去除的含硫杂质。
[0080]
应用对比例2本应用对比例中,按照应用例5中的步骤,进行乙醛偶姻缩合反应制备乙偶姻。与应用例5 的区别仅在于,将ph由8换成7。
[0081]
经检测,本应用对比例中,乙醛转化率为42.6%,乙偶姻选择性为60.9%,乙偶姻总收率为25.9%。
[0082]
结果分析:相较于应用例5而言,应用对比例2中乙醛转化率和乙偶姻产物的收率明显较低,推测原因在于:当反应体系的ph过低时,会导致催化活性物种中的卡宾碳位点难
以去质子化,导致催化活性下降,进而影响乙醛偶姻缩合反应的速率,大量乙醛未被转化。
[0083]
应用对比例3本应用对比例中,按照应用例4中的步骤,进行乙醛偶姻缩合反应制备乙偶姻。与应用例4 的区别仅在于,将反应温度由60℃换成40℃。
[0084]
经检测,本应用对比例中,乙醛转化率为22.9%,乙偶姻选择性为51.5%,乙偶姻总收率为11.8%。
[0085]
结果分析:相较于应用例4而言,应用对比例3中乙醛转化率和乙偶姻产物的收率明显较低,推测原因在于:当反应温度过低时,催化活性物种中的卡宾碳位点难以去质子化,会造成催化剂的催化活性过低,进而影响乙醛偶姻缩合反应的速率。
[0086]
应用对比例4本应用对比例中,按照应用例5中的步骤,进行乙醛偶姻缩合反应制备乙偶姻。与应用例5 的区别仅在于,将反应温度由150℃换成170℃。
[0087]
经检测,本应用对比例中,乙醛转化率为68.2%,乙偶姻选择性为19.6%,乙偶姻总收率为13.4%。
[0088]
结果分析:相较于应用例5而言,应用对比例4中乙偶姻产物的选择性和收率明显较低,推测原因在于:当温度过高时,非均相催化剂中的噻唑环会发生开环分解,不仅会造成催化剂的乙醛偶姻缩合反应催化活性和选择性下降,同时还会产生较多难以去除的含硫杂质。
[0089]
本发明中所用原料、设备,若无特别说明,均为本领域的常用原料、设备;本发明中所用方法,若无特别说明,均为本领域的常规方法。
[0090]
以上所述,仅是本发明的较佳实施例,并非对本发明作任何限制,凡是根据本发明技术实质对以上实施例所作的任何简单修改、变更以及等效变换,均仍属于本发明技术方案的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。