1.本发明属化学分析技术领域,涉及氮杂-氟硼二吡咯甲川在制备测定临界胶束浓度的荧光指示剂中的应用,尤其是氮杂-氟硼二吡咯甲川化合物在制备测定两亲性表面活性物质临界胶束浓度中的应用。
背景技术:
2.现有技术中,两亲性化合物指分子结构中一端亲水、一端亲油的分子化合物,视亲水/亲油基团大小或链长不同,以及基团亲水性和亲油性的程度不同,具有不同程度的表面活性作用。通常所指的表面活性剂即为表面活性作用相对较强的两亲性化合物,在化工、医药、材料、生物等领域具有广泛的应用,其用途涉及分散、增溶、乳化、润湿、发泡、消沫、润滑、洗涤、抗静电、涂层等多个方面。近年来,随着聚合物科学的不断发展,产生了系列两亲性嵌段聚合物。其中部分作为表面活性剂已被广泛使用,如聚氧乙烯-聚氧丙烯嵌段共聚物(常见商品名为poloxamer或pluronic);部分被用作药物递送的载体,如聚乳酸-聚乙二醇嵌段共聚物(peg-pla)。此外,尚有为数众多的弱表面活性两亲性化合物,它们存在于天然产物中,如植物皂苷等,也可以通过合成、半合成的手段获得。
3.研究显示,两亲性表面活性物质在水中具有一定的溶解度,但当浓度增加达到某一阀值时,两亲性分子会自发聚集形成疏水端朝向内核、亲水端朝向外水相的热力学稳定胶束结构;胶束开始形成时的两亲性化合物浓度,即该化合物的临界胶束浓度(critical micelle concentration,cmc)。在cmc之下,两亲性化合物以单体形式存在于相界面或溶解于水相中;在cmc之上,两亲性化合物分子同时存在于相界面、水相和胶束结构中;由于胶束的形成,两亲性化合物溶液体系的多种物理化学性质将发生显著的变化,如增溶作用的增强、表面活性的削弱、光学性质的变化等。cmc通常可以认为是表面活性物质物理化学性质的分水岭。在表面活性物质的实际应用中以及两亲性化合物的研制开发过程中,测定cmc 值具有非常现实的指导意义。
4.cmc值是表面活性剂等两亲性表面活性物质质量或性能评价的重要指标之一,也是分析化学、物理化学、材料化学领域的研究热点之一。已用于实践或已报道的cmc测定方法种类繁多,但无一例外地,均是利用当浓度达到cmc时表面活性物质溶液的物理或化学性质的突变来确定cmc值的。根据所选定的突变指标的性质,cmc测定法大致可分成两大类:第一类为物理参数法,单纯利用表面活性物质在形成胶束前后物理性质的变化来确定cmc值,可利用的物理参数主要包括表面张力、电导率、电位、光谱、浊度、光散射、粘度等;第二类为溶质法,藉胶束形成对特定溶质的增溶作用,通过测定溶质在cmc上下物理化学性质的突变来测定cmc值,可利用的参数包括溶解度、可见-紫外光谱、荧光光谱等。然而,目前并没有法定的cmc值测定法。实际操作中,往往视表面活性物质的性质 (包括预估的cmc值)、测定环境的要求、仪器设备的限制等选择适当的测定方法。各种cmc测定法具有各自的适用范围与局限性,文献报道的表面活性剂的 cmc值往往差异也非常大。表1列举了常见cmc测定法的
指示指标、适用范围及其优缺点,表2列举了见诸报道的常见表面活性剂的cmc值测定结果差异。分析背景资料发现,第一类物理参数法往往需要特殊的检测仪器;由于物理参数测定本身灵敏度不高,往往造成该类方法灵敏度不佳,只适合于mm水平的cmc测定;尤其是在cmc附近,突变终点不易确认,准确度也因此受影响;此外,某些特定方法只适合于特定表面活性物质的测定,如电导法只适用于可解离的离子型表面活性物质。第二类利用溶质性质突变的测定法,视溶质突变信号的强弱其灵敏度也不同,当采用突变信号十分强烈的指示剂,如荧光指示剂时,其灵敏度大大加强,从而可以满足μm甚至nm水平的cmc值测定的需要。
5.表1为常见cmc测定法的指示指标、适用范围及优缺点:
[0006][0007][0008]
表2为文献报道的常见表面活性剂cmc测定值:
[0009][0010]
[0011]
荧光探针法是目前cmc值测定最为常用的方法,相对于其他测定法,其优点主要包括灵敏度高、操作方便、成本低、可视化等,采用通用型荧光分光光度计即可进行。荧光探针法以荧光探针为指示剂,通过测定表面活性物质的浓度在达到cmc时(即胶束形成时)荧光性质的突变来确定cmc测定终点。其基本原理在于荧光探针对表面活性物质体系中的不同相环境(水相和胶束内核疏水相)具有不同的响应性。一般可利用的荧光突变信号主要包括荧光发射波长和荧光强度,也可利用荧光光谱特征峰的比值或衍生参数实现信号放大,如芘荧光测定法即是利用i
339
/i
333
或者i1/i3的荧光强度比值来进行突变终点判定的,其中,荧光强度是最常被利用的荧光突变判定指标;图1为典型的荧光探针测定法cmc 突变终点判定示意图,通常选择cmc上、下规律性比较明显的各点分别作线性回归,两条直线相交点对应的浓度即为cmc。
[0012]
尽管多种荧光探针已被应用于cmc测定的实践中,或者被报道用于实验过程中cmc值的测定。但经分析发现,各种荧光探针在测定cmc的应用中均存在或多或少的缺陷,大大限制了其应用,目前,荧光探针法存在的问题如下所述: (1)荧光突变不明显,测定终点难以判定;造成该情况的主要原因是所采用的荧光指示剂对环境的响应性不强,导致荧光突变不显著,cmc突变点上下的规律性不强,不易判定终点;另一方面,如果荧光指示剂的发射强度不足,往往需要消耗较大量的荧光指示剂并且在较高胶束浓度下才能准确测定,但在cmc浓度附近因荧光强度太低而难以准确确定终点。(2)cmc测定终点判定变异大,导致测定精密度不佳;通常在cmc之上,随着胶束浓度的不断增加,所溶解荧光探针的浓度也不断增加,因此荧光强度也随之增加,当达到一定浓度后,荧光强度与表面活性物质浓度之间的线性比较容易确定,并且十分稳定;但是,在 cmc之下,胶束尚未形成,荧光探针以极低浓度分散于水相中,荧光测定变异系数较大,线性往往不佳,从而影响测定精密度。(3)光不稳定性。某些结构类别的荧光探针,具有一定程度的光不稳定性,在光照情况下会发生光淬灭或光漂白效应,从而干扰测定,并对避光操作提出了较高的要求,操作十分不便。(4) 灵敏度不够;一般的荧光探针可满足常见cmc在mm和μm水平的表面活性物质的测定要求,仅有极少数的荧光探针可满足nm水平cmc值的测定;两亲性嵌段聚合物的cmc值通常在nm水平,随着其不断发展,对cmc测定法也提出了更高的要求。
[0013]
中国发明专利cn103411961b、cn104193666b、cn106546564b、cn109738402a、 cn108387559a、cn110028446a公开了以聚集诱导发光(aggregation-inducedemission,aie)有机化合物为荧光指示剂测定表面活性剂cmc的应用和方法。 aie荧光探针溶于良溶剂以及胶束内核中时不发光,分散于外水相中迅速聚集沉淀,因分子内运动受限而致发光(angew chem int ed engl 2020.doi: 10.1002/anie.202007525;angew chem int ed engl 2020,59(25):9888-907; chem commun 2014,50(9):1107-9;analyst 2011,136(16):3343-8;chem soc rev 2011,40(11):5361-88.)。利用aie荧光探针的这一性质,将一定浓度的aie探针与不同浓度的表面活性剂溶液以一定的方式相混合,荧光强度突变的最强点即为cmc。但由于受聚集体沉淀物性质的影响,突变区域的荧光测定常常受到干扰,对于突变最强点的确定往往并不准确,这大大影响了测定的准确度。受各种因素的影响,aie荧光探针法仅适用于cmc在100μm以上的表面活性剂体系。上述中国发明专利公开中仅提供了十二烷基硫酸钠(sds)、十六烷基三甲基溴化铵 (ctab)等少数几种表面活性剂cmc测定的实施例。显然,上述中国发明专利所公开的方法并不适用于cmc在nm水平的两亲性嵌段聚
合物cmc的测定,在灵敏度、准确度、精确度、适用范围方面均无法和本发明的应用和方法相比。
[0014]
中国发明专利cn104897628b公开了发射波长在紫外-可见光区的新结构荧光探针用于测定常见表面活性剂cmc的应用,所述荧光探针法不具备本发明所述应用具有的准确度、灵敏度和稳定性。采用导数法确定终点不能够真实反映胶束形成的起始点,即真正的cmc。该发明专利公开中仅给出了阴离子表面活性剂十二烷基硫酸钠和非离子型表面活性剂曲拉通x-100的cmc测定实施例。
[0015]
中国发明专利cn109270035b公开了采用n-b
18h22
为荧光探针物质测定十二烷基硫酸钠cmc的方法,其原理是基于十二烷基硫酸钠单体可以线性增强n-b
18h22
荧光探针的荧光强度,伴随着胶束的形成,荧光增强被削弱,通过荧光突变可以测定cmc。该方法仅适用于十二烷基硫酸钠一种表面活性剂cmc的测定。
[0016]
基于现有技术的现状,本技术的发明人拟提供涉及氮杂-氟硼二吡咯甲川在制备测定临界胶束浓度的荧光指示剂中的应用,尤其是氮杂-氟硼二吡咯甲川化合物在制备测定两亲性表面活性物质临界胶束浓度的荧光指示剂中的应用。
技术实现要素:
[0017]
本发明的目的在于基于现有技术的现状,提供利用具有氮杂-氟硼二吡咯甲川类化合物为荧光指示剂测定表面活性物质cmc值的应用;具体涉及氮杂-氟硼二吡咯甲川在制备测定临界胶束浓度的荧光指示剂中的应用,尤其是氮杂-氟硼二吡咯甲川化合物在制备测定两亲性表面活性物质临界胶束浓度的荧光指示剂中的应用。
[0018]
本发明基于,理想的荧光探针应具有很高的灵敏度和很好的准确度、精确度,要求所采用的荧光探针具有良好的化学与光稳定性、灵敏的环境响应性和高量子产率;最理想的荧光探针在cmc之下应无荧光,对测定不形成干扰,并且突变显著,有助于准确判定测定终点等基础,采用具有氮杂-氟硼二吡咯甲川母核结构的荧光探针用于表面活性物质cmc值的测定,结果显示,以氮杂-氟硼二吡咯甲川化合物作为荧光指示剂测定cmc,与传统荧光指示剂相比,具有灵敏度、准确度、精确度俱佳的优势,尤其是在两亲性嵌段聚合物nm水平cmc值的测定方面;更特别地,本发明所述的氮杂-氟硼二吡咯甲川化合物具有在表面活性物质cmc以下无荧光的特征,作为其能保证其应用灵敏度、准确度、精密度俱佳的最主要因素,并且因此可简化荧光探针法测定cmc的操作步骤。
[0019]
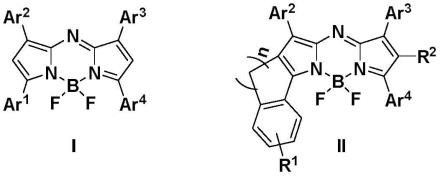
本发明所述的氮杂-氟硼二吡咯甲川化合物是具有式(i)或(ii)基本结构的化合物。
[0020][0021]
本发明所述氮杂-氟硼二吡咯甲川化合物在制备测定两亲性表面活性物质临界胶束浓度的荧光指示剂中的应用中,依据的原理是:所述荧光指示剂在两亲性表面活性物质
cmc以下水溶液中受疏水作用力的驱动,形成聚集体,基于聚集导致淬灭(aggregation-caused quenching,acq)效应而致荧光淬灭,在cmc以上当胶束形成时,所述荧光指示剂能够分配进入胶束内核,以单分子激发状态存在,从而发射荧光,并且荧光强度与胶束浓度或表面活性物质浓度(cmc以上) 呈正相关,通过检测荧光强度突变点可测得cmc值。相对于传统的荧光指示剂,本发明所述具有式(i)或(ii)氮杂-氟硼二吡咯甲川结构的荧光化合物在浓度低于cmc的表面活性物质的水溶液中,其荧光完全淬灭,对测定无干扰,使得对突变终点的判定更为准确;
[0022]
本发明中,因所述具有式(i)或(ii)氮杂-氟硼二吡咯甲川结构的荧光化合物用于测定nm水平的两亲性嵌段共聚物cmc值时,具有很高的响应值,而传统荧光指示剂无法用于测定nm水平的cmc值或测定不准确;本发明所述具有式 (i)或(ii)氮杂-氟硼二吡咯甲川结构的荧光化合物比传统荧光指示剂具有更好的化学和光学稳定性。
[0023]
除此之外,由于本发明的具有式(i)或(ii)氮杂-氟硼二吡咯甲川结构的荧光化合物在浓度低于cmc的表面活性物质的水溶液中完全无荧光,因此在确定 cmc测定终点时,不需要对cmc以下各点进行线性回归操作,根据突变区域趋势线的切线与浓度轴的交点直接读取,或者cmc以上线性区域的回归直线与浓度轴的截距计算,即可测得cmc值,简化了操作。
[0024]
综合上述特点,本发明所述的氮杂-氟硼二吡咯甲川化合物在制备测定两亲性表面活性物质临界胶束浓度的荧光指示剂中的应用,比现有技术的荧光探针法具有更好的准确性、精密度、灵敏性和稳定性,适用范围也更加广泛,操作也更为简便。
[0025]
在本发明的一些实施方案中,
[0026]
ar1、ar2、ar3、ar4选自苯基、c
1-6
烷氧基取代的苯基、c
1-6
烷基取代的苯基、甲基哌嗪基取代的苯基、羟基取代的苯基、吡啶基、噻吩基、c
1-6
烷基取代的噻吩基、呋喃基、c
1-6
烷基取代的呋喃基;
[0027]
n选自1、2;
[0028]
r1选自h、c
1-6
烷氧基、c
1-6
烷基;
[0029]
r2选自h或任选与ar4形成五元或六元并环结构。
[0030]
在本发明的一些优选的实施方案中,
[0031]
ar1、ar2、ar3、ar4选自苯基、甲氧基取代的苯基、甲基取代的苯基、甲基哌嗪基取代的苯基、羟基取代的苯基、吡啶基、噻吩基、甲基取代的噻吩基、呋喃基、甲基取代的呋喃基;
[0032]
和/或
[0033]
r1选自h、甲基、甲氧基;
[0034]
和/或
[0035]
r2选自h或任选与ar4形成五元或六元并环结构;当r2与ar4形成五元或六元并环结构时,ar4为苯基或取代的苯基。
[0036]
在本发明中,取代基可在苯基、杂芳基上任意可取代的位置取代,如邻位、间位、对位。
[0037]
更具体地,本发明所示具有式(i)或(ii)氮杂-氟硼二吡咯甲川结构的荧光化合物的化学结构及其在乙腈(ch3cn)溶剂中的基本荧光参数(φ为荧光量子产率,λ
abs
为最大吸收波长,ε为摩尔消光系数,λ
em
为最大发射波长)如下所示:
[0038]
[0039]
[0040]
[0041]
[0042][0043]
本发明中,所示的氮杂-氟硼二吡咯甲川类荧光探针,最大吸收波长在 624-755nm之间,最大荧光发射波长在679-776nm之间,属于近红外区域;绝大多数当前常用的cmc测定用荧光探针其最大发射波长小于600nm。随着测定波长红移,光散射与背景自发荧光减弱,因此对cmc测定干扰减少。本发明所示的氮杂-氟硼二吡咯甲川类荧光探针,量子产率高,乙腈溶液中介于0.10-0.41 之间,优选化合物1-15的量子产率介于0.21-0.35之间。较高的量子产率意味着使用较少的荧光探针就可以获得较高的响应信号,有助于提高cmc测定的灵敏度。本发明所述的氮杂-氟硼二吡咯甲川类荧光探针化学性质稳定,光学性质也十分稳定,无光淬灭和光漂白现象,操作时不需要避光,如实施例6中所示,以化合物2为荧光指示
剂,样品放置24个小时,荧光响应信号强度与cmc测定结果基本保持不变。相比较而言,传统荧光探针如本发明对比例中的dph、hoechst 33342等易受测定环境中光的影响,导致荧光探针发生降解、异构化等变化,通常在测定操作中要求避光,非常不方便。
[0044]
本发明所述应用的具体实施可采取以下后孵育法、前孵育法、稀释法三种方式中的任意一种或多种方式的操作步骤或其变化步骤。后孵育法的特点是事先配制一系列浓度的表面活性物质水溶液,浓度高于cmc的体系会自发形成胶束,荧光指示剂在胶束形成后加入并与表面活性物质的水溶液共孵育,荧光指示剂分子分配至胶束内核而发光。前孵育法与此恰恰相反,荧光指示剂在胶束形成之前加入至表面活性物质中,在加水水化形成胶束的过程中荧光指示剂分配进入胶束内核而发光。稀释法的特点是事先配制表面活性物质的高浓度溶液与荧光指示剂的混合体系,荧光指示剂分配与胶束内核而发光,伴随着表面活性物质体系的稀释过程,荧光强度随表面活性物质浓度降低而等比例降低,当浓度降低至cmc以下时,胶束完全解聚,荧光指示剂也随之分散至水环境中,导致荧光淬灭,据此测定cmc。具体步骤说明如下:
[0045]
后孵育法:
[0046]
步骤1.将式(i)或(ii)所示结构化合物用易挥发性良溶剂配制成储备溶液;
[0047]
步骤2.配制包括cmc在内的一系列浓度的表面活性物质水溶液,低于cmc 和高于cmc浓度的数量应足以保证绘制具有明显突变点的荧光强度与表面活性剂物质浓度曲线图;
[0048]
步骤3.取等体积步骤1中储备液加入等体积的步骤2中系列浓度表面活性物质溶液中,混匀,挥去溶剂,静置孵育,室温至少8小时或者37℃至少4小时,在最大发射波长处测定荧光强度,绘制荧光强度-表面活性物质浓度曲线。对突变区域趋势线作最大斜率切线,以切线与浓度轴的交点读取cmc值;或将 cmc以上线性区域各点进行线性回归,根据与浓度轴的截矩直接计算cmc值。
[0049]
以上后孵育法的步骤适用于所有类型表面活性物质cmc的测定,包括但不限于阴离子表面活性剂、阳离子表面活性剂、非离子表面活性剂、两亲性嵌段共聚物。
[0050]
所述后孵育法步骤的变化是:(1)步骤3中取等体积步骤1中储备液加入敞口容器中,挥去溶剂;(2)步骤3中分别取等体积步骤2中系列浓度表面活性物质溶液,加入变化步骤(1)中敞口容器中,混匀,静置孵育,室温至少8小时或者37℃至少4小时,在最大发射波长处测定荧光强度,绘制荧光强度-表面活性物质浓度曲线。对突变区域趋势线作最大斜率切线,最大斜率切线定义为cmc 突变区域趋势线斜率最大的高浓度区域切线,本发明中但凡涉及最大斜率切线概念,均与此处意义相同。以切线与浓度轴的交点读取cmc值。或者将cmc以上线性区域各点进行线性回归,根据与浓度轴的截矩直接计算cmc值。
[0051]
以上后孵育法的变化步骤适用于所有类型表面活性物质cmc的测定,包括但不限于阴离子表面活性剂、阳离子表面活性剂、非离子表面活性剂、两亲性嵌段共聚物。
[0052]
上述操作方法的要点是事先配制一系列浓度的表面活性物质的水溶液,然后加入氮杂-氟硼二吡咯甲川化合物的储备液,混匀,或者与除去溶剂的荧光探针混合均匀,孵育一定时间,以使荧光探针分子充分进入胶束的疏水内核。由于所述操作便捷,本发明的实施例中均给出了采用上述后孵育法测定cmc的步骤。实施例1中进一步具体说明,将化合物1的浓度为34μm的乙腈储备液,分别加入一系列浓度的非离子表面活性剂曲拉通x-100的溶液中,荧光发射光谱(如图2 所示)显示,在630-800nm波长范围内,低于0.2mm以下,完全无荧
光;0.2mm 以上,荧光强度随曲拉通x-100的浓度增加而增强;以荧光强度对曲拉通x-100 的浓度作图(如图3所示),对突变趋势线作最大斜率切线,因cmc以下荧光值为零,通过切线与浓度轴的截距即可测得cmc为0.19mm;所述的步骤适用于所有表面活性物质cmc的测定,本发明的实施例3、4中给出了以化合物1为荧光指示剂,应用上述步骤测定阴离子表面活性剂sds(如图5所示)、两嵌段聚合物mpeg
5k-pdlla
5k
(如图6所示)的cmc测定结果判定图,最终测得的cmc值分别为7.7mm、186nm。对于曲拉通x-100,选择cmc以上线性区域内曲拉通x-100 浓度分别为0.20、0.25、0.30及0.40mm的各点进行线性回归,得直线方程为 y=1154c-223.11,相关系数r2为0.9769,直线在浓度轴的截距为0.19mm,即为其cmc;对于sds,选择cmc以上线性区域内浓度分别为8、9及10mm的各点进行线性回归,得线性方程为y=55.23c
–
409.75,相关系数r2为0.9995,直线在浓度轴的截距为7.70mm,即为其cmc;对于mpeg
5k-pdlla
5k
,选择线性区域内浓度分别为0.4、0.6、0.8、1.0μm的各点对荧光值与浓度的对数做线性回归,得线性方程为y=39.216c 28.231,相关系数r2为0.9993,基于截距法得到该材料的cmc为191nm。对比切线法与线性回归法判定cmc值,两种操作方法所得结果十分接近;切线法的优点在于操作简便,只要获得明显的趋势线即可绘制最大斜率切线,不足之处是cmc值是在浓度轴上读取获得的,受绘制最大斜率切线变异的影响,数据常有波动。线性回归法操作更为简便,cmc值的确定是计算得到的,变异较小,不足之处是为了获得足够多的数据点进行线性回归常常要求在相应区域设置更多浓度点;一般情况下,最大斜率切线与回归直线比较接近;理想情况下,最大斜率切线与回归直线能够达到高度的重合。
[0053]
鉴于所采取后孵育法步骤中用于溶解荧光指示剂的有机溶剂用量有可能对 cmc的测定形成干扰,并且难以完全挥发除去,操作过程中应控制其用量,在满足准确量取体积情况下以用量最少为最佳。在本发明实施例所示后孵育法步骤中,以有机溶剂溶解的荧光指示剂储备液与预先配制的表面活性物质系列浓度溶液的体积比为1:200进行混合,有机溶剂的影响可忽略不计;本发明通过对比实施例1中不除溶解荧光指示剂的溶剂与事先去除溶剂的操作所得结果得以验证;本发明所示后孵育法操作步骤条件下事先去除或后加不除有机溶剂所测得的曲拉通x-100的cmc均为0.19mm。
[0054]
前孵育法:
[0055]
步骤1.将式(i)或(ii)所示结构化合物用易挥发性良溶剂配制成储备溶液;
[0056]
步骤2.用易挥发性良溶剂配制包括cmc在内的一系列浓度的表面活性物质储备液,低于cmc和高于cmc浓度的数量应足以保证绘制具有明显突变点的荧光强度-表面活性物质浓度曲线图;
[0057]
步骤3.分别取等体积步骤2中储备液加入敞口容器中,再分别加入等体积步骤1中储备液,混匀,挥去溶剂,加入等体积的水,振摇,静置孵育,室温至少8小时或者37℃至少4小时,在最大发射波长处测定荧光强度,绘制荧光强度-表面活性物质浓度曲线。对突变区域趋势线作最大斜率切线,以切线与浓度轴的交点读取cmc值;或将cmc以上线性区域各点进行线性回归,根据与浓度轴的截矩直接计算cmc值。
[0058]
以上前孵育法步骤适用于所有类型表面活性物质cmc的测定,包括但不限于阴离子表面活性剂、阳离子表面活性剂、非离子表面活性剂、两亲性嵌段共聚物,尤其适用于两亲性嵌段共聚物cmc值的测定。
[0059]
所述前孵育法步骤的变化是:(1)步骤2中将待测表面活性物质用易挥发性良溶液
配制成高浓度的储备液;(2)步骤3中分别取不同体积的变化步骤(1) 中储备液置不同的敞口容器中,再分别加入等体积的前孵育法步骤1中储备液,混匀,挥去溶剂,加入等体积的水,振摇,静置孵育,室温至少8小时或者37℃至少8小时,在最大发射波长处测定荧光强度,绘制荧光强度-表面活性物质浓度曲线。对突变区域趋势线作最大斜率切线,以切线与浓度轴的交点读取cmc 值;或将cmc以上线性区域各点进行线性回归,根据与浓度轴的截矩直接计算 cmc值。
[0060]
所述前孵育法的变化步骤适用于所有类型表面活性物质cmc的测定,包括但不限于阴离子表面活性剂、阳离子表面活性剂、非离子表面活性剂、两亲性嵌段共聚物,尤其适用于两亲性嵌段共聚物cmc值的测定。
[0061]
稀释法:
[0062]
步骤1.将式(i)或(ii)所示结构化合物用易挥发性良溶剂配制成储备溶液;
[0063]
步骤2.配制高于cmc的表面活性物质水溶液;
[0064]
步骤3.将一定体积步骤1中储备液直接加入步骤2中表面活性物质水溶液中,混匀,挥去溶剂;或将一定体积步骤1中储备液先加入敞口容器中,挥去溶剂,再加入步骤2中表面活性物质溶液,混匀后于37℃下孵育12小时,并冷却至室温;
[0065]
步骤4.步骤3高浓度溶液中依次加入一定体积水进行稀释,直到cmc以下,测定荧光强度,绘制荧光强度-表面活性物质实际浓度的曲线图。对突变区域趋势线作最大斜率切线,以切线与浓度轴的交点读取cmc值;或将cmc以上线性区域各点进行线性回归,根据与浓度轴的截矩直接计算cmc值。
[0066]
上述稀释法步骤适用于所有类型表面活性物质cmc的测定,包括但不限于阴离子表面活性剂、阳离子表面活性剂、非离子表面活性剂、两亲性嵌段共聚物。
[0067]
本发明中,所述后孵育法、前孵育法、稀释法所示步骤或者变化步骤中,所涉及采用有机溶剂配制荧光指示剂溶液的步骤,其目的是将荧光指示剂完全溶解并配制成所需浓度的溶液,以便定量取样或加入至表面活性物质的溶液中;同样地,前孵育法中涉及采用有机溶剂溶解表面活性物质配制成溶液以便与荧光指示剂溶液相混合的步骤;所采用有机溶剂不参与胶束的形成过程或者荧光指示剂向胶束内核的迁移分配过程,与cmc的测定过程无直接关系;实施过程中,应尽可能除去;因此,应选择对荧光指示剂以及表面活性物质具有很好溶解性能并能够很容易除去的有机溶剂,所述良溶剂为乙腈、二氯甲烷、甲醇、乙醇、丙醇、异丙醇、正丁醇、异丁醇、叔丁醇、丙酮、乙醚、乙酸乙酯、三氯甲烷、四氢呋喃、二甲亚砜、n,n-二甲基甲酰胺中的一种或几种的混合溶剂。
[0068]
本发明中,所述后孵育法、前孵育法、稀释法所示步骤或者变化步骤中,所述表面活性物质溶液为水溶液,并非特指纯水的溶液体系,由于本发明所示氮杂
ꢀ‑
氟硼二吡咯甲川结构化合物本身的化学与光学性质并不受常见电解质、水中可以自由溶解的溶质的影响,因此本发明所述应用同样适用于两亲性表面活性物质的盐溶液、缓冲液,以及含其他水中自由溶解溶质如葡萄糖等常见糖类物质的体系。
[0069]
本发明中,所述后孵育法、前孵育法、稀释法所示步骤或者变化步骤中,荧光强度的测定在本发明各氮杂-氟硼二吡咯甲川结构化合物的最大激发与最大发射波长条件下进行,以获得最大的信号响应。在偏离最大激发与最大发射波长条件下测定荧光强度,信号响应值会降低,但在一定范围内对cmc测定结果没有影响。本发明实施例中均是在荧光指示剂
的最大激发和最大发射波长处测定荧光强度,但是这并不对本发明的应用形成限定。
[0070]
本发明中,所述后孵育法、前孵育法、稀释法所示步骤或者变化步骤中,在将荧光探针与表面活性物质以不同的方式混合后,孵育操作是必须的步骤,其作用是保证荧光探针能够充分分配到胶束的内核并达到平衡,或者是在胶束的平衡被打破后再次达到平衡,外在表现是所测得的荧光强度达到一个稳定值。孵育时间对于达平衡至关重要,通常常用荧光指示剂需要孵育12-24小时,如本发明对比实施例1、2、10中以芘、hoechst 33342、dph为荧光指示剂测定均需要孵育至少12小时以上。而以本发明的所有化合物为荧光指示剂,孵育时间可以大大缩短,室温条件下,孵育8小时即可;如果采用37℃条件下孵育4小时,待冷却至室温后测定荧光强度,即可获得稳定的荧光强度测定结果。
[0071]
本发明中,所述后孵育法、前孵育法、稀释法所示步骤或者变化步骤中,cmc 测定终点的确定是通过绘制荧光强度与表面活性物质实际浓度的曲线图,对突变区域趋势线作最大斜率切线或对cmc以上线性区域进行回归,以直线与浓度轴的交点读取或以浓度轴截矩计算cmc值。本发明的实施例中均给出了cmc测定终点的判定方式,即以突变区域趋势线的切线与浓度轴(y=0)的交点读取,或者以线性区域回归直线与浓度轴的截距计算获得。以实施例1来进一步说明,化合物 1的荧光强度-曲拉通x-100的曲线图中,低浓度区域荧光值为零,因此不需要进行任何操作,以cmc以上突变区域趋势线的切线与浓度轴交点读取曲拉通 x-100的cmc值为0.19mm;以cmc以上曲拉通x-100浓度为0.20、0.25、0.30 及0.40mm的各点作线性回归,通过与浓度轴的截距求得曲拉通x-100的cmc 值亦为0.19mm。本发明所示cmc测定终点判定方法操作简便,并且排除了因cmc 以下数据波动而带来的干扰,明显减少了测定误差。
[0072]
鉴于本发明氮杂-氟硼二吡咯甲川化合物量子产率高,荧光信号响应强,再配合选择最大激发和发射波长处测定,可最大程度地提高信号响应值。因此,只需要采用极少量的荧光指示剂就可以实施本发明的应用。如实施例中所示,对于 cmc较高的体系(曲拉通x-100、sds、ctab等),氮杂-氟硼二吡咯甲川荧光指示剂的初始浓度为34μm,以1:200的比例加入表面活性物质溶液中;对于cmc 较低的体系(两亲性嵌段聚合物mpeg-pdlla等),氮杂-氟硼二吡咯甲川荧光指示剂的初始浓度为3.4μm,以1:200的比例加入表面活性物质溶液中,即可实施本发明的应用。而对比例1中芘和对比例2中hoechst 33342则需要起始浓度设定为400μm,以1:200的比例加入表面活性物质溶液中,高于本发明氮杂
‑ꢀ
氟硼二吡咯甲川荧光指示剂浓度有十倍之多。对比例3中,tpe的初始浓度需要 100μm,对比例10中荧光指示剂dph由于量子产率较高,则需要初始浓度为50 μm。本发明实施例中所采用的氮杂-氟硼二吡咯甲川荧光指示剂的浓度是为了更好地说明本发明的应用优势,并不对本发明的应用形成限定。
[0073]
本发明所述氮杂-氟硼二吡咯甲川化合物相对于传统荧光指示剂具有的优势包括:测定终定更容易判断,准确度更高,精密度更好。这主要得益于本发明的惊喜发现,即所述氮杂-氟硼二吡咯甲川化合物全部具有完全的聚集导致淬灭 (acq)效应,cmc下水溶液中无荧光的特征,本发明实施例所提供的cmc测定终点判定图中均明确地说明了这一特征。与此相反,以对比例1中芘、对比例2 中hoechst 33342、对比例3中aie荧光染料tpe、对比例10中dph、对比例4-9 中氟硼二吡咯甲川化合物(对比化合物1-3)为荧光指示剂,不具有完全acq效应,cmc以下仍然具有荧光。在cmc突变终点附近,尤其是cmc以下低浓度区域,测定值
波动较大,影响测定的准确度,多次重复测定精密度不佳。
[0074]
本发明为充分展示所述氮杂-氟硼二吡咯甲川化合物荧光指示剂的适用范围,选择表2中不同性质的常用表面活性剂作为实施例,主要包括:阴离子表面活性剂sds、阳离子表面活性剂ctab、非离子表面活性剂曲拉通x-100和吐温 80,以及两亲性嵌段共聚物泊洛沙姆、dspe-peg2000、不同嵌段链长的 mpeg-pdlla共聚物。本发明为充分展示所述氮杂-氟硼二吡咯甲川化合物荧光指示剂相对于传统荧光指示剂的优势,选择代表性荧光染料芘、hoechst 33342、 dph、aie类荧光探针tpe,以及不具有cmc以下荧光完全淬灭效应的氟硼二吡咯甲川化合物类荧光染料作为对比例。
[0075]
本发明实施例1中,将化合物1配制成34μm的乙腈储备液,分别按照后孵育法或前孵育法步骤操作,配制一系列浓度介于0.01-2mm的曲拉通x-100 水溶液。将探针储备液以1:200(v/v)比例加入至系列浓度的曲拉通x-100溶液中,37℃下孵育4小时,待冷却至室温后,或者室温孵育8小时后,测定荧光。化合物1在浓度低于0.2mm的曲拉通x-100水溶液中,荧光发射光谱(附图2) 显示在630-800nm波长范围内完全无荧光,化合物1因其acq效应完全无信号。随着曲拉通x-100浓度逐渐增加至0.2mm及以上时,化合物1荧光开始出现且逐渐增强。以最大激发/发射波长处的荧光值对曲拉通x-100浓度作图(附图3),结果表明各数据点的分布符合s型曲线,这是由于随着曲拉通x-100浓度的增加,曲拉通x-100由单体分子逐步自组装形成胶束,溶液中的化合物1由未包载状态逐步变为部分包载、最终转变为完全包载状态,荧光信号也呈逐渐增强趋势。因 cmc以下荧光值为零,相关数据不需要进行任何操作。仅需要对突变区域趋势线作最大斜率切线,通过与浓度轴(y=0)的截距即可测得cmc为0.19mm。荧光突变点判定不受cmc以下低浓度区域波动的影响。cmc以上高浓度区,荧光值测定相对稳定,受干扰较少。因此,测定准确度得以保证,重复性较好。选择cmc 以上线性区域内曲拉通x-100浓度分别为0.20、0.25、0.30及0.40mm的各点进行线性回归,得直线方程为y=1154c-223.11,相关系数r2为0.9769,直线在浓度轴的截距为0.19mm,即为曲拉通x-100得cmc值。
[0076]
本发明实施例2中给出了采用化合物1连续测定5次曲拉通x-100cmc值的结果,分别为0.19、0.19、0.20、0.20、0.19mm,标准差为2.5%,精密度非常好(附图4)。
[0077]
同样地,以化合物1为荧光指示剂,实施例3、4给出了测定阴离子表面活性剂十二烷基硫酸钠(附图5)、两亲性嵌段聚合物mpeg
5k-pdlla
5k
(附图6)的测定结果判定图,最终测得的cmc值分别为7.7mm、186nm。无一例外地,在 cmc以下化合物1均无荧光。测定精密度均很好。
[0078]
本发明所述实施例1-4中,化合物1的初始浓度在测定cmc在微摩尔浓度水平的曲拉通x-100和cmc在毫摩尔浓度水平的sds的cmc值时设定为34μm,即可获得很好的信号响应和测定结果重现性。对于cmc在纳摩尔浓度水平的两亲性嵌段聚合物mpeg
5k-pdlla
5k
,信号突跃区域附近胶束浓度低,胶束内核无法容纳大量的荧光指示剂。因此,化合物1的初始浓度设定为3.4μm,由于化合物1 量子产率较高,并且测定干扰能够被有效排除,在此浓度下即可实现cmc的准确测定。而对于量子产率较低的荧光指示剂,必须使用较高的剂量才能够获得较好的信号响应,而过高的荧光指示剂用量提示仅在较高胶束浓度下才能够准确测定荧光信号,在cmc附近突变区域反而无法产生较为强烈的荧光信号,因此也就无法准确地指示cmc的突变。
[0079]
本发明实施例5、6、7、8中,采用化合物2分别对曲拉通x-100、mpeg
5k-pdlla
5k
的cmc进行了测定,并评价了其测定cmc的稳定性与重复性。
[0080]
所述实施例5中测定了化合物2在不同浓度的曲拉通x-100溶液中的荧光光谱(附图7),随着曲拉通x-100浓度的逐渐降低,荧光发射强度也逐渐降低,在0.2mm浓度以下,荧光强度降低为几乎为零。与化合物1相同,该现象是基于完全的acq效应,而传统低波长荧光探针不具有完全的acq效应。按后孵育法或前孵育法步骤或其变化步骤进行测定,以荧光强度对曲拉通x-100浓度作图 (附图8),对突变区域趋势线上各浓度点作切线,在浓度轴(y=0)上读取cmc 值均为0.20mm。化合物2的程度结果与化合物1略有区别,这可能是由于测定偶然误差造成的。采用稀释法步骤,将化合物2配制成85μm的乙腈储备液,按照1:200(v/v)的比例加入至0.4mm的曲拉通x-100的水溶液中,用去离子水稀释成系列稀溶液,静置一段时间后测定荧光强度,以荧光强度对曲拉通x-100 浓度作图(附图9),对突变趋势线各点作切线,在浓度轴上读取cmc值分别为 0.19mm(静置1小时)和0.20mm(静置4小时);同样用化合物2的水溶液进行稀释,测得的cmc值为0.21mm(静置1小时)和0.20mm(静置4小时)(附图10)。静置时间对cmc测定有一定影响,足够的静置时间(至少4小时)有利于稀释后探针分子在胶束内外相之间达到平衡,趋于稳定状态,从而保证测定的准确性。
[0081]
所述实施例6中,采用后孵育法或前孵育法,进一步评价了长时间暴露在光和氧气条件下,化合物2测定曲拉通cmc的稳定性。研究结果表明,暴露在光、氧等不利条件下,各样品中化合物2的荧光无显著变化(附图11),测得的cmc 分别为0.20、0.19及0.20mm,标准差为2.9%。这里仅以化合物2为代表评价其稳定性,事实上,本发明的所有氮杂-氟硼二吡咯甲川化合物都具有很好的化学与光学稳定性。
[0082]
所述实施例7中,按前孵育法的步骤对mpeg
5k-pdlla
5k
的cmc进行测定,化合物2在不同浓度的共聚物溶液中的发射光谱如附图12,在0.1μm以下几乎无荧光,以荧光强度对共聚物浓度作图,测得cmc结果为0.19μm(附图13)。同一实施例中,采用后孵育法,测得的荧光发射光谱如附图14,以荧光强度对共聚物浓度作图,测得cmc结果为0.19μm(附图15)。两种不同的操作步骤测得的结果相同。无论是使用前孵育法还是后孵育法,都不显著影响采用本发明氮杂
ꢀ‑
氟硼二吡咯甲川化合物测定两亲性嵌段聚合物cmc值的结果。
[0083]
所述实施例8中,以化合物2为荧光指示剂,采用后孵育法多批次测定了 mpeg
5k-pdlla
5k
的cmc,结果分别为0.10、0.10及0.10μm,标准差为0.7%,说明方法重复性好(附图16)。化合物2的测定结果与化合物1相同,进一步说明了本发明的应用和方法的适用性和可靠性。
[0084]
本发明实施例9、10、11、12中,采用化合物3分别对曲拉通x-100、sds、 ctab、mpeg
5k-pdlla
5k
的cmc进行了测定。
[0085]
所述实施例9中,对化合物3在不同浓度曲拉通x-100溶液中的荧光发射光谱进行了测定,如附图17所示,在浓度低于0.2mm的曲拉通x-100水溶液中几乎无荧光。同化合物1和2,其原理是基于完全的acq效应。按后孵育法步骤或其变化步骤进行测定,以荧光强度对曲拉通x-100浓度作图(附图18),对突变趋势线各点作切线,在浓度轴上读取cmc值均为0.20mm。测定结果与化合物1 的测定结果完全相同,与化合物2的测定结果略有不同。如上所述,这可能是由于测定偶然误差造成的。
[0086]
所述实施例10中,以化合物3为荧光指示剂,分别按照后孵育法或前孵育法及其变
化步骤对sds的cmc进行测定,结果为7.4mm(附图19)。化合物3 的测定结果与化合物1的测定结果基本一致,进一步说明了本发明的应用与方法在不同氮杂-氟硼二吡咯甲川化合物之间的适用性。
[0087]
所述实施例11中,以化合物3为荧光指示剂,分别按照后孵育法或前孵育法及其变化步骤对阳离子表面活性剂ctab的cmc进行测定,结果为0.80mm(附图20)。说明本发明的应用和方法除了上述阴离子表面活性剂、非离子表面活性剂、两亲性聚合物,同样适用于阳离子表面活性剂。
[0088]
所述实施例12中,以化合物3为荧光指示剂,按照后孵育法及其变化步骤对mpeg
5k-pdlla
5k
的cmc进行测定,结果为0.19μm(附图21)。化合物3的测定结果与化合物1和2的测定结果完全一致,说明本发明的应用与方法在两亲性嵌段共聚物cmc测定中的适用性良好。
[0089]
本发明的实施例13中,分别以化合物4-57为荧光指示剂,分别对sds、ctab、曲拉通x-100的cmc按照后孵育法或者前孵育法及其变化步骤进行测定,结果表明,对于同一表面活性剂,各化合物得到的cmc测定结果基本保持一致,仅有个别化合物的结果发生偏离。这说明本发明的cmc测定结果在不同化合物的荧光探针中得以验证,充分说明了本发明所依据测定原理的适用性与可靠性。
[0090]
本发明的实施例14中,以化合物1、2、3为荧光指示剂分别对更多表面活性剂吐温80、吐温20、聚氧乙烯氢化蓖麻油el、聚氧乙烯氢化蓖麻油elp、苄泽58、苄泽59、麦泽52的cmc进行了测定。采用不同荧光指示剂测得的同一表面活性剂的cmc值重现性非常好,尽管存在一定的差异,但并不显著。充分说明本发明的应用和方法具有推广应用于其他更多种类表面活性物质cmc值测定的潜力。
[0091]
本发明的实施例15中,以化合物1、2、3为荧光指示剂,分别对更多具有不同嵌段类型和链长的两亲性嵌段共聚物mpeg
5k-pdlla
3k
、mpeg
2.5k-pdlla
2.5k
、 dspe-peg2000、泊洛沙姆407、普朗尼克p104,以及接枝共聚物聚乙烯己内酰胺
ꢀ‑
聚乙酸乙烯酯-聚乙二醇接枝共聚物(soluplus)的cmc进行了测定。采用不同荧光指示剂测得的同一两亲性嵌段共聚物或接枝共聚物的cmc值重现性非常好,尽管存在一定的差异,但并不显著。充分说明本发明的应用和方法推广应用于其他更多种类具有不同嵌段类型和链长的两亲性嵌段共聚物或接枝共聚物cmc值测定的潜力。
[0092]
本发明的实施例16中,描述了以化合物1、2、3为荧光指示剂测定普朗尼克p104临界胶束温度(cmt)的操作与结果。cmt指在特定浓度下,两亲性化合物随温度变化形成胶束的临界温度。其原理与操作步骤和cmc测定基本类同,所不同的是本实施例中cmt的测定观察指标是荧光强度随温度变化而发生突变的情况。以化合物2为例,具体地按本发明后孵育法类似步骤操作。先行配制化合物2的乙腈储备液(34μm)和普朗尼克p104的水溶液(0.5mg/ml,w/v)。将化合物2的储备液按照1:200(v/v)的比例加入至普朗尼克p104水溶液中,混匀,置于5、10、15、20、25、30、35及40℃的恒温水浴中,控制水浴温度(
±ꢀ
0.5℃)。超声10分钟后,检测各样品的荧光。附图22为不同温度下样品中化合物2的荧光光谱,当温度低于普朗尼克p104的cmt时,p104处于单分子状态,难以包载化合物2,化合物2由于完全的acq效应而完全没有荧光;而当温度高于cmt时,普朗尼克p104逐渐形成胶束而包载化合物2,在突变范围内,胶束浓度与温度呈正相关,化合物2的荧光随温度升高而逐渐增加直至稳定。以最大激发和发射波长处测得的荧光值对温度的响应曲线见附图23。在25-30℃温度区间,荧光发
生突变,突变点指示为cmt。通过对突变趋势线作最大斜率切线,与温度轴相交点可读取cmt。本实施例中,0.5mg/ml的普朗尼克p104的cmt测得为27.7℃。同样地,以化合物1和化合物3为荧光指示剂测得0.5mg/ml的普朗尼克p104的cmt分别为27.2℃和27.9℃。实施例16作为举例仅展示了以化合物1、2、3测定p104特定浓度的cmt的操作与结果,但是这并不限定本发明的应用。本发明应用用于测定cmt的范畴可涵盖本发明所有权利要求的氮杂-氟硼二吡咯甲川类化合物、更多两亲性化合物,以及更多两亲性化合物的更多浓度。
[0093]
本发明的对比例1中描述了采用芘为荧光指示剂测定cmc的步骤与测定结果。由于芘是当前最为常用的荧光指示剂之一,本发明中以其作为对比例来说明本发明氮杂-氟硼二吡咯甲川类化合物作为荧光指示剂的优点。芘荧光cmc测定法最常用的响应指标主要有:
①
芘的荧光发射光谱(固定激发波长335nm)的第一峰值i1(372nm)和第三峰值i3(384nm)的比值(i1/i3)的突变(附图 24);
②
以372nm为固定发射波长,得到的荧光激发光谱中i
339
和i
333
的比值 (i
339
/i
333
)的突变(附图25)。如突变终点判定图(附图26、27)所示,在突变终点附近,尤其是cmc值以下,响应指标波动非常大,这直接影响了对测定终点的判定。以i1/i3为指标测定曲拉通x-100(附图26)、吐温80(附图27)的cmc 分别为0.25mm和15.3μm,而以i
339
/i
333
为指标测得两种表面活性剂的cmc值分别为0.23mm(附图26)和4.66μm(附图27)。同样采用芘为指示剂,以两种不同指标测定的结果都存在着很大的差异。以i
339
/i
333
为指标连续测定三组吐温80样品的cmc分别为6.17、5.45及3.47mm,标准差为27.8%(附图28),精密度不佳。
[0094]
本发明的对比例2中给出了以常用荧光染料hoechst 33342为指示剂测定 cmc的步骤与结果。hoechst 33342在水中具有浓度淬灭效应,导致荧光发射减弱,但不同于本发明的氮杂-氟硼二吡咯甲川类化合物,hoechst 33342在水中的荧光不会完全消失,而本发明的氮杂-氟硼二吡咯甲川化合物在水中是完全没有荧光的。以hoechst 33342为指示剂测定cmc的原理与本发明中氮杂-氟硼二吡咯甲川化合物有相似之处,都属于在cmc之上荧光增强型荧光指示剂。所不同的是,本发明的氮杂-氟硼二吡咯甲川化合物在cmc之下荧光绝对淬灭,荧光强度降低为零,而hoechst 33342在cmc之下仍然具有荧光。附图29为hoechst 33342在不同浓度的曲拉通x-100溶液中的荧光光谱图,随着表面活性物质浓度的增加,染料分子分配到胶束内核中,荧光随之增强。附图30中展示了以hoechst 33342为指示剂测定曲拉通x-100cmc的结果判定图,结果表明在cmc突变点以下,hoechst 33342仍具有荧光,并且荧光测定值有较大的变异,这大大影响了对突变终点的准确判定。通过对突变趋势线作最大斜率切线,与低浓度区域直线拟合相交点,读取cmc值为0.25mm,显著大于采用本发明以氮杂-氟硼二吡咯甲川化合物为指示剂测定的结果。很显然,本发明应用的结果判定准确度与精密度显著优于传统染料hoechst 33342。对比例2仅仅给出了一种典型的荧光增强型常用染料用于测定cmc的示范,事实上所有常用的荧光染料在测定cmc时都有类似的情况出现。除本发明的氮杂-氟硼二吡咯甲川化合物外,都具有cmc以下有荧光的情况。
[0095]
本发明的对比例3中给出了以具有aie性质的tpe荧光染料为指示剂测定 cmc的测定步骤与结果。aie类荧光染料与本发明化合物的荧光响应趋势相反。aie荧光染料在cmc以下发射荧光,但在cmc以上,分配至胶束内核中时,荧光减弱或消失。如附图31所示,通过后孵育法步骤测定cmc,发现tpe在不同浓度的曲拉通x-100溶液中的荧光强度随着曲拉通浓度的增加而逐渐降低,在高于曲拉通x-100cmc(0.20mm)时,最大发射波长处的荧光强度大
幅度降低。在最大发射波长450nm处测定荧光强度,对曲拉通x-100浓度作图(附图32),得到的荧光强度发生突变的初始点对应浓度为0.25mm,即为曲拉通x-100的 cmc。由于该方法得到的cmc对应的浓度为曲拉通胶束完全包载tpe的浓度,因此所得结果往往偏高于其真实cmc。此外,在曲拉通x-100浓度低于其cmc时, tpe虽有较强荧光,但波动较大。而当曲拉通x-100浓度高于其cmc时,tpe即使被完全包载,仍会保留一部分荧光。这种由强变弱的趋势不易于准确计算cmc 值。基于稀释法得到的对十二烷基硫酸钠cmc的测定结果如附图33所示,tpe 在不同浓度sds条件下,荧光强度缓慢上升,根本不存在荧光突变,无法按荧光突变最强点判定cmc(chem commun 2014,50(9):1107-9;cn103411961b、 cn104193666b)。上述结果说明,tpe在cmc以下形成聚集体,通常以沉淀形式存在,其荧光强度受聚集体物理形态的影响较大,因此测定值变异大。附图34 中,考察了不同浓度的tpe加入水中形成聚集体沉淀后,荧光强度的变化情况,结果表明,tpe聚集体的浓度与其荧光强度的线性较差,并且变异系数大,这可能是造成以这类aie荧光染料为指示剂测定cmc突变不明显,终点不易判定的最主要原因。
[0096]
本发明的对比例4中给出了以芘为荧光指示剂以i
339
/i
333
为突变指标测定两亲性嵌段共聚物mpeg
5k-pdlla
5k
的cmc的结果判定图(附图35)。三次测定结果分别为1.18、0.90及0.39μm,标准差为48.8%,精确度不佳。本发明的氮杂
‑ꢀ
氟硼二吡咯甲川化合物用于测定两亲性共聚物mpeg
5k-pdlla
5k
的cmc,精密度显著优于传统的芘荧光指示剂。
[0097]
本发明的对比例5-9为利用具有与本发明氮杂-氟硼二吡咯甲川化合物相似结构的氟硼二吡咯甲川化合物为荧光指示剂测定代表性表面活性物质cmc的步骤与结果,用于与本发明的氮杂-氟硼二吡咯甲川化合物类荧光指示剂进行比较,以说明本发明应用的优越性。
[0098]
所述对比例5中,给出了以基本结构为氟硼二吡咯甲川的对比化合物1为荧光指示剂测定曲拉通x-100的cmc的步骤和结果,具体步骤可采用本发明所示后孵育法、前孵育法或稀释法步骤或者其变化步骤进行,但在对比例5中,仅以后孵育法步骤举例说明。首先将对比化合物1配制成34μm的乙腈储备液,以及浓度介于0.005-0.75mm的曲拉通x-100水溶液。将对比化合物1的储备液以 1:200(v/v)比例加入至系列浓度的曲拉通x-100溶液中,混匀,37℃下孵育4 小时以上,冷却至室温,待测。以537nm为固定激发波长,对比化合物1在不同浓度的曲拉通x-100溶液中的荧光发射光谱如附图36所示。对比化合物1在各浓度曲拉通x-100溶液中均有较强的发射荧光。从最大发射波长571nm处的荧光值对曲拉通x-100浓度曲线图(附图37)可以看出,随着曲拉通x-100浓度的增加,对比化合物1的荧光信号有逐渐下降趋势,呈现一定程度的响应性,但突变并不明显,因此无法准确读取cmc值。本发明的氮杂-氟硼二吡咯甲川化合物在所有被检测两亲性活性物质的cmc以下的水溶液中均没有荧光,并且随着两亲性表面活性物质在水溶液中的浓度增加,而发生显著突跃,cmc测定更为准确,可排除对比化合物1因突变终点难以判断而到来的测定干扰。
[0099]
所述对比例6中,给出了以基本结构为氟硼二吡咯甲川的对比化合物2为荧光指示剂测定曲拉通x-100的cmc的步骤和结果,具体步骤可采用本发明所示后孵育法、前孵育法和稀释法步骤或者其变化步骤进行,但在对比例6中,仅以后孵育法步骤举例说明。首先将对比化合物2配制成34μm的乙腈储备液,以及浓度介于0.005-0.75mm的曲拉通x-100水溶液。将对比化合物1的储备液以 1:200(v/v)比例加入至系列浓度的曲拉通x-100溶液中,混
匀,37℃下孵育4 小时以上,冷却至室温,待测。以564nm为固定激发波长,各样品中对比化合物2的荧光发射光谱如附图38所示。无论样品中曲拉通x-100的浓度高于cmc 抑或低于cmc,对比化合物2一直具有荧光。以最大发射波长处(610nm)的荧光值对曲拉通x-100浓度作曲线,结果见附图39。当曲拉通x-100浓度低于cmc 时,对比化合物2的荧光较弱,但随着曲拉通浓度增加,对比化合物2的荧光逐渐增强;当曲拉通x-100浓度增加至其cmc以上时,对比化合物2的荧光增强更为明显。由于对比化合物2在曲拉通x-100浓度高于或低于cmc时皆具有荧光,荧光变化的转折点并不明显。并且,在曲拉通x-100浓度低于cmc时对比化合物 2的荧光已经呈现逐渐增强的趋势,这极容易被误认为此时曲拉通x-100已经开始形成胶束。通过切线法得到曲拉通x-100的cmc值为0.156mm,与文献报道的0.20mm有较大的差距。
[0100]
所述对比例7中,给出了以基本结构为氟硼二吡咯甲川的对比化合物2为荧光指示剂测定sds的cmc的步骤和结果,具体步骤可采用本发明所示后孵育法、前孵育法和稀释法步骤或者其变化步骤进行,但在对比例7中,仅以后孵育法步骤举例说明。首先将对比化合物2配制成34μm的乙腈储备液,以及浓度介于 0.15-20mm的sds水溶液。将对比化合物2的储备液以1:200(v/v)比例加入至各sds溶液中,混匀,37℃下孵育4小时以上,冷却至室温,待测。以564nm 为固定激发波长,各样品中对比化合物2的荧光发射光谱如附图40所示。无论 sds浓度高于或低于其cmc,化合物2始终具有较强的荧光信号。以最大发射波长(610nm)的荧光值对sds浓度作图,结果见附图41。荧光强度随sds浓度的变化呈单调增强的趋势。图中无法观察到明显的突变点,无法测得cmc值。
[0101]
所述对比例8中,给出了以基本结构为氟硼二吡咯甲川的对比化合物3为荧光指示剂测定曲拉通x-100的cmc的步骤和结果,具体步骤可采用本发明所示后孵育法、前孵育法和稀释法步骤或者其变化步骤进行,但在对比例6中,仅以后孵育法步骤举例说明。首先将对比化合物3配制成34μm的乙腈储备液,以及浓度介于0.005-0.75mm的曲拉通x-100水溶液。将对比化合物2的储备液以 1:200(v/v)比例加入至各曲拉通x-100溶液中,混匀,37℃下孵育4小时以上,冷却至室温,待测。以562nm为固定激发波长,各样品中对比化合物3的荧光发射光谱如附图42所示。无论样品中曲拉通x-100的浓度高于cmc抑或低于cmc,对比化合物3一直具有荧光。以最大发射波长处(608nm)的荧光值对曲拉通 x-100浓度作曲线,结果见附图43。当曲拉通x-100浓度低于cmc时,对比化合物3的荧光已经出现且逐渐增强;当曲拉通x-100浓度高于其cmc时,对比化合物3的荧光增强更为明显。由于对比化合物3在曲拉通x-100浓度高于或低于 cmc时皆具有荧光,荧光变化的转折点并不明显。并且,在曲拉通x-100浓度低于cmc时对比化合物3的荧光已经呈现逐渐增强的趋势,这极容易被误认为此时曲拉通x-100已经开始形成胶束。通过切线法得到的cmc值为0.153mm,与文献报道的0.20mm有较大的差距。
[0102]
所述对比例9中,给出了以基本结构为氟硼二吡咯甲川的对比化合物3为荧光指示剂测定sds的cmc的步骤和结果,具体步骤可采用本发明所示后孵育法、前孵育法和稀释法步骤或者其变化步骤进行,但在对比例9中,仅以后孵育法步骤举例说明。首先将对比化合物3配制成34μm的乙腈储备液,以及浓度介于 0.15-20mm的sds水溶液。将对比化合物3的储备液以1:200(v/v)比例加入至各曲拉通x-100溶液中,混匀,37℃下孵育4小时,冷却至室温,待测。以 562nm为固定激发波长,各样品中对比化合物3的荧光发射光谱如附图44所示。无论sds浓度高于或低于其cmc,对比化合物3始终具有较强的荧光信号。以最大发射波长
(608nm)的荧光值对sds浓度作图,结果见附图45。荧光强度随 sds浓度的变化呈单调增强的趋势。与其他bodipy探针相似,同样无法从图中观察到明显的突变点,并且无法通过切线法对sds的cmc值进行计算。
[0103]
所述对比例10中,给出了以常用荧光探针dph为荧光指示剂测定 dspe-peg2000的cmc的步骤和结果,具体步骤以后孵育法步骤举例说明。将dph 配制成50μm的四氢呋喃储备液,分别配制浓度为0.04、0.02、0.07、0.18、 0.36、0.71、1.4、2.1、2.9、3.6、7.1、14.3、35.7μm的dspe-peg2000水溶液。将dph储备液按1:200(v/v)比例加入至系列浓度的dspe-peg2000水溶液中,室温孵育12小时,测定各样品中dph的荧光。以355nm为固定激发波长,得到各样品中dph的荧光发射光谱。如附图46所示,无论样品中dspe-peg2000 的浓度高于cmc抑或低于cmc,dph一直具有较强的荧光。以最大发射波长处的荧光值对dspe-peg2000浓度作图,如附图47所示。当dspe-peg2000浓度低于其cmc时,dph的荧光具有较大的波动性,对测定终点得判定造成一定得干扰。通过切线法得到的第一个交点对应的浓度为1.1μm,大于其他方法得到的cmc 值。
附图说明
[0104]
图1典型的荧光探针法测定cmc突变终点判定示意图。
[0105]
图2化合物1在不同浓度曲拉通x-100水溶液中的荧光发射光谱图。
[0106]
图3化合物1荧光强度相对于曲拉通x-100浓度的变化趋势图。
[0107]
图4化合物1荧光强度相对于曲拉通x-100浓度的多组变化趋势图。
[0108]
图5化合物1荧光强度相对于sds浓度的变化趋势图。
[0109]
图6化合物1荧光强度相对于两亲性共聚物mpeg
5k-pdlla
5k
浓度的变化趋势图。
[0110]
图7化合物2在不同浓度曲拉通x-100水溶液中的荧光发射光谱图。
[0111]
图8化合物2荧光强度相对于曲拉通x-100浓度的变化趋势图。
[0112]
图9静置不同时间条件下化合物2荧光强度相对于曲拉通x-100浓度的变化趋势图(以去离子水稀释)。
[0113]
图10静置不同时间条件下化合物2荧光强度相对于曲拉通x-100浓度的变化趋势图(以化合物2的水溶液稀释)。
[0114]
图11不同时间点下化合物2荧光强度相对于曲拉通x-100浓度的变化趋势图。图12高浓度化合物2(34μm)在不同浓度两亲性共聚物mpeg
5k-pdlla
5k
水溶液中的荧光发射光谱图。
[0115]
图13高浓度化合物2(34μm)荧光强度相对于两亲性共聚物mpeg
5k-pdlla
5k
浓度的变化趋势图。
[0116]
图14低浓度化合物2(3.4μm)在不同浓度两亲性共聚物mpeg
5k-pdlla
5k
水溶液中的荧光发射光谱图。
[0117]
图15低浓度化合物2(3.4μm)荧光强度相对于两亲性共聚物mpeg
5k-pdlla
5k
浓度的变化趋势图。
[0118]
图16化合物2荧光强度相对于曲拉通x-100浓度的多组变化趋势图。
[0119]
图17化合物3在不同浓度曲拉通x-100水溶液中的荧光发射光谱图。
[0120]
图18化合物3荧光强度相对于曲拉通x-100浓度的变化趋势图。
[0121]
图19化合物3荧光强度相对于sds浓度的变化趋势图。
[0122]
图20化合物3荧光强度相对于ctab浓度的变化趋势图。
[0123]
图21化合物3荧光强度相对于两亲性共聚物mpeg
5k-pdlla
5k
浓度的变化趋势图。
[0124]
图22化合物2在不同温度普朗尼克p104(0.5mg/ml)水溶液中的荧光发射光谱图。
[0125]
图23化合物2在普朗尼克p104(0.5mg/ml)水溶液中的荧光强度相对于温度的变化趋势图。
[0126]
图24以335nm为固定激发波长不同浓度的芘荧光探针的发射光谱图(360-450 nm)。
[0127]
图25以371nm为固定发射波长不同浓度的芘荧光探针的激发光谱图(300-360 nm)。
[0128]
图26芘荧光探针法测定曲拉通x-100cmc突变指标i
339
/i
333
和i1/i3相对于浓度的变化趋势图。
[0129]
图27芘荧光探针法测定tween 80cmc突变指标i
339
/i
333
和i1/i3相对于浓度的变化趋势图。
[0130]
图28芘荧光探针法测定tween 80cmc突变指标i
339
/i
333
相对于浓度的变化趋势图(三批)。
[0131]
图29荧光探针hoechst 33342在不同浓度曲拉通x-100水溶液中的荧光发射光谱图。
[0132]
图30 hoechst 33342荧光强度相对于曲拉通x-100浓度的变化趋势图。
[0133]
图31 aie荧光探针tpe在不同浓度曲拉通x-100水溶液中的荧光发射光谱图。
[0134]
图32 tpe荧光强度相对于曲拉通x-100浓度的变化趋势图。
[0135]
图33 tpe荧光强度相对于sds浓度的变化趋势图。
[0136]
图34 tpe荧光强度随水中浓度变化趋势图。
[0137]
图35芘荧光探针法测定两亲性共聚物mpeg
5k-pdlla
5k cmc三批测定结果判定变化趋势图。
[0138]
图36对比化合物1在不同浓度曲拉通x-100水溶液中的荧光发射光谱图。
[0139]
图37对比化合物1荧光强度相对于曲拉通x-100浓度的变化趋势图。
[0140]
图38对比化合物2在不同浓度曲拉通x-100水溶液中的荧光发射光谱图。
[0141]
图39对比化合物2荧光强度相对于曲拉通x-100浓度的变化趋势图。
[0142]
图40对比化合物2在不同浓度sds水溶液中的荧光发射光谱图。
[0143]
图41对比化合物2荧光强度相对于sds浓度的变化趋势图。
[0144]
图42对比化合物3在不同浓度曲拉通x-100水溶液中的荧光发射光谱图。
[0145]
图43对比化合物3荧光强度相对于曲拉通x-100浓度的变化趋势图。
[0146]
图44对比化合物3在不同浓度sds水溶液中的荧光发射光谱图。
[0147]
图45对比化合物3荧光强度相对于sds浓度的变化趋势图。
[0148]
图46dph在不同浓度dspe-peg2000水溶液中的荧光发射光谱图。
[0149]
图47dph荧光强度相对于dspe-peg2000浓度的变化趋势图。
具体实施方式
[0150]
以下结合实施例来进一步解释本发明,但实施例并不对本发明做任何形式的限定。
[0151]
实施例1以化合物1为荧光指示剂测定曲拉通x-100的cmc
[0152]
将化合物1配置成34μm的乙腈储备液,分别配制浓度为0.01、0.02、0.05、 0.08、
0.1、0.2、0.25、0.3、0.4、0.5、0.6、0.8、1.0、1.5、2.0mm的曲拉通x-100水溶液。将探针储备液以1:200(v/v)比例加入至系列浓度的曲拉通 x-100溶液中,37℃下孵育4小时,待冷却至室温后,测定荧光。得到的发射光谱见附图2,当曲拉通x-100浓度小于0.2mm时,化合物2因其聚集导致淬灭效应完全无信号。随着曲拉通x-100浓度逐渐增加至0.2mm及以上时,化合物 2荧光开始出现且逐渐增强。以最大激发、发射波长处的荧光值对曲拉通x-100 浓度作图,结果见附图3。各数据点的分布符合s型曲线,这是由于随着曲拉通 x-100浓度的增加,曲拉通x-100由单体分子逐步自组装形成胶束,溶液中的化合物2由无包载状态逐步变为部分包载、最终转为完全包载状态。对s型曲线做切线,得到的第一个交点对应的浓度为0.19mm,即为曲拉通x-100的cmc值。选择cmc以上线性区域内曲拉通x-100浓度分别为0.20、0.25、0.30及0.40mm 的各点进行线性回归,得直线方程为y=1154c-223.11,相关系数r为0.9769,直线在浓度轴的截距为0.19mm,即为曲拉通x-100得cmc值。
[0153]
将上述化合物1的浓度为34μm的乙腈储备液置于试管中,吹干溶剂,分别加入上述系列浓度的曲拉通x-100水溶液,混匀,孵育4小时,以下按同样方法操作,测得曲拉通x-100的cmc值为0.22mm,与上述不除溶剂的操作步骤所测得的cmc值完全一致。
[0154]
实施例2以化合物1为荧光指示剂多批次测定曲拉通x-100的cmc
[0155]
将化合物1配置成34μm的乙腈储备液,分别配制浓度为0.01、0.02、0.05、 0.08、0.1、0.2、0.25、0.3、0.4、0.5、0.6、0.8、1.0、1.5、2.0mm的曲拉通x-100水溶液。将探针储备液以1:200(v/v)比例加入至各曲拉通x-100溶液中。平行操作5份,37℃下孵育4小时,待冷却至室温后,测定荧光。以最大激发、发射波长处的荧光值对曲拉通x-100浓度作图,结果见附图4。各组数据点的分布皆符合s型曲线,且相互之间基本上完全重叠。对s型曲线突变区域做切线,得到的与浓度轴的交点对应的浓度分别为0.19、0.19、0.20、0.20及0.19 mm,标准差为2.5%,重复性好。
[0156]
实施例3以化合物1为荧光指示剂测定sds的cmc
[0157]
将化合物1配制成34μm的乙腈储备液,分别配制浓度分别为0.1、0.5、 1、3、4、5、6、7、8、9、10、15、20mm的sds水溶液。将探针储备液以1:200 (v/v)比例加入至各sds溶液中,37℃下孵育4小时以上,待冷却至室温后,测定荧光。以最大发射波长荧光值对sds浓度作图,结果见附图4。荧光强度随sds浓度的变化符合s型曲线,与化合物1在不同浓度sds溶液中的包载状态一致。对s型曲线分别做切线,得到的第一个交点对应的浓度为7.6mm,即为sds 的cmc值。
[0158]
实施例4以化合物1为荧光指示剂测定mpeg
5k-pdlla
5k
的cmc
[0159]
将化合物1配制成3.4μm的乙腈储备液,分别配制浓度为0.001、0.005、 0.01、0.02、0.05、0.1、0.2、0.4、0.6、0.8、1.0、2.0、4.0、10.0的mpeg
5k-pdlla
5k
的水溶液。将探针储备液以1:200(v/v)比例加入至各溶液中,37℃下孵育4 小时以上,待冷却至室温后,测定荧光。得到的化合物1的荧光强度随 mpeg
5k-pdlla
5k
浓度变化曲线见附图6,当mpeg
5k-pdlla
5k
浓度小于0.1μm时,化合物1因其在水溶液中聚集而完全无信号。随着mpeg
5k-pdlla
5k
浓度逐渐增加至0.1mm及以上时,化合物1荧光开始出现且逐渐增强。各数据点的分布同样极其符合s型曲线。对s型曲线分别做切线,得到的第一个交点对应的浓度为 0.09μm,即为mpeg
5k-pdlla
5k
的cmc值。
[0160]
实施例5以化合物2为荧光指示剂测定曲拉通x-100的cmc
[0161]
将化合物2配制成34μm的乙腈储备液,分别配制浓度为0.01、0.02、0.05、 0.08、0.1、0.2、0.25、0.3、0.4、0.5、0.6、0.8、1.0、1.5、2.0mm的曲拉通x-100水溶液。将探针储备液以1:200(v/v)比例加入至系列浓度的曲拉通 x-100溶液中,37℃下孵育4小时以上,待冷却至室温后,测定荧光。得到的发射光谱见附图7,当曲拉通x-100浓度小于0.2mm时,化合物2因其聚集导致淬灭效应完全无信号。随着曲拉通x-100浓度逐渐增加至0.2mm及以上时,化合物2荧光开始出现且逐渐增强。以最大激发、发射波长处的荧光值对曲拉通 x-100浓度作图,结果见附图8。各数据点的分布极其符合s型曲线,这是由于随着曲拉通x-100浓度的增加,曲拉通x-100由单体分子逐步自组装形成胶束,溶液中的化合物2由无包载状态逐步变为部分包载、最终转为完全包载状态。对 s型曲线分别做切线,得到的第一个交点对应的浓度为0.20mm,即为曲拉通 x-100的cmc值。
[0162]
将化合物2配制成85μm的乙腈储备液,按照1:200(v/v)比例加入至0.4 mm的曲拉通x-100水溶液中,37℃下孵育12小时后,冷却至室温。随即,分别用去离子水或化合物2的水溶液稀释曲拉通x-100溶液至0.36、0.32、0.28、 0.24、0.20、0.16、0.10及0.08mm。静置一段时间后,测定各样品中化合物2 在最大激发与发射波长处的荧光值,并以荧光值对曲拉通x-100浓度做曲线。经去离子水稀释,所得到的曲线见附图9。随着曲拉通x-100浓度的下降,化合物 2的荧光线性降低直至消失。取荧光消失的初始点对应的浓度为cmc,得cmc为 0.19mm(静置1小时)与0.20mm(静置4小时)。经化合物2水溶液稀释,所得到的曲线见附图10。同样取荧光消失的初始点对应的浓度为cmc,得cmc为 0.21mm(静置1小时)与0.20mm(静置4小时)。两种稀释方法得到的cmc值相同,且不受静置时间的影响。
[0163]
实施例6以化合物2为荧光指示剂于不同时间点多次测定曲拉通x-100 的cmc
[0164]
将化合物2配制成34μm的乙腈储备液,分别配制浓度为0.01、0.02、0.05、 0.08、0.1、0.2、0.25、0.3、0.4、0.5、0.6、0.8、1.0、1.5、2.0mm的曲拉通x-100水溶液。将探针储备液以1:200(v/v)比例加入至系列浓度的曲拉通 x-100溶液中,37℃下孵育4小时以上,随即冷却至室温。随后分别在室温下(无避光、无避氧)静置0、12及24小时,测定样品中化合物2在最大激发与发射波长下的荧光强度。并以其荧光强度对曲拉通x-100的浓度做曲线,结果见附图 11。不同时间点测得的化合物2的荧光强度无显著变化,拟合的s型曲线基本上完全重叠。基于线性回归得到的曲拉通x-100的cmc分别为0.20、0.19及0.20 mm,标准差为2.9%,方法的精密度好。
[0165]
实施例7以化合物2为荧光指示剂测定mpeg
5k-pdlla
5k
的cmc
[0166]
将化合物2配制成3.4μm的乙腈储备液,分别配制浓度为0.001、0.005、 0.01、0.02、0.05、0.1、0.2、0.4、0.6、0.8、1.0、2.0、4.0、10.0的mpeg
5k-pdlla
5k
乙腈溶液。将探针储备液以1:200(v/v)比例加入至各溶液中,采用空气挥干有机溶剂,形成材料薄膜。随即加入等体积的水,37℃下孵育4小时以上,待冷却至室温后,测定荧光。得到的发射光谱见附图12,当mpeg
5k-pdlla
5k
浓度小于0.1μm时,化合物2因其在水溶液中聚集而完全无信号。随着mpeg
5k-pdlla
5k
浓度逐渐增加至0.1μm及以上时,化合物2荧光开始出现且逐渐增强。以最大激发、发射波长处的荧光值对材料浓度作图,结果见附图13。各数据点的分布同样极其符合s型曲线。对s型曲线分别做切线,得到的与浓度轴的交点对应的浓度为0.09μm,即为mpeg
5k-pdlla
5k
的cmc值。
[0167]
将化合物2配制成3.4μm的乙腈储备液,分别配制浓度为0.001、0.005、 0.01、
0.02、0.05、0.1、0.2、0.4、0.6、0.8、1.0、2.0、4.0、10.0的mpeg
5k-pdlla
5k
水溶液。将探针储备液以1:200(v/v)比例加入至各溶液中,37℃下孵育4小时以上,待冷却至室温后,测定荧光。得到的荧光发射光谱见附图14,当 mpeg
5k-pdlla
5k
浓度小于0.1μm时,化合物2无荧光信号。随着mpeg
5k-pdlla
5k
浓度逐渐增加至0.1μm及以上时,化合物2荧光出现且逐渐增强。以最大激发、发射波长荧光值对材料浓度作图,结果见附图15。对s型曲线做切线,得到的与浓度轴的交点对应的浓度为0.10μm,即为mpeg
5k-pdlla
5k
的cmc值。
[0168]
实施例8以化合物2为荧光指示剂多批次测定mpeg
5k-pdlla
5k
的cmc
[0169]
将化合物2配制成3.4μm的乙腈储备液,分别配制浓度为0.001、0.005、 0.01、0.02、0.05、0.1、0.2、0.4、0.6、0.8、1.0、2.0、4.0、10.0的mpeg
5k-pdlla
5k
水溶液。将探针储备液以1:200(v/v)比例加入至各溶液中。平行配制3份,并于37℃下孵育4小时以上,待冷却至室温后,测定荧光。以最大激发、发射波长处的荧光值对材料浓度作图,结果见附图16。对各s型曲线分别做切线,得到的mpeg
5k-pdlla
5k
的cmc分别为0.10、0.10、0.10μm,标准差为0.7%,精密度高。
[0170]
实施例9以化合物3为荧光指示剂测定曲拉通x-100的cmc
[0171]
将化合物3配制成34μm的乙腈储备液,分别配制浓度为0.01、0.02、0.05、 0.08、0.1、0.2、0.25、0.3、0.4、0.5、0.6、0.8、1、1.5、2mm的曲拉通x-100 水溶液。将探针储备液以1:200(v/v)比例加入至各曲拉通x-100溶液中,37℃下孵育4小时以上,待冷却至室温后,测定荧光。得到的荧光发射光谱见附图 17,当曲拉通x-100浓度小于0.2mm时,化合物3因其聚集导致淬灭效应完全无信号。随着曲拉通x-100浓度逐渐增加至0.2mm及以上时,化合物3荧光开始出现且逐渐增强。以最大发射波长荧光值对曲拉通x-100浓度作图,结果见附图18。各数据点的分布极其符合s型曲线,这是由于随着曲拉通x-100浓度的增加,曲拉通x-100由单体分子逐步自组装形成胶束,溶液中的化合物3由无包载状态逐步变为部分包载、最终转为完全包载状态。对s型曲线分别做切线,得到的第一个交点对应的浓度为0.22mm,即为曲拉通x-100的cmc值。
[0172]
实施例10以化合物3为荧光指示剂测定sds的cmc
[0173]
将化合物3配制成34μm的乙腈储备液,分别配制浓度为0.1、0.5、1、3、 4、5、6、7、8、9、10、15、20mm的sds水溶液。将探针储备液以1:200(v/v) 比例加入至各sds溶液中,37℃下孵育4小时以上,待冷却至室温后,测定荧光。以最大发射波长荧光值对sds浓度作图,结果见附图19。荧光强度随sds浓度的变化符合s型曲线,与化合物3在不同浓度sds溶液中的包载状态一致。对s 型曲线分别做切线,得到的第一个交点对应的浓度为7.7mm,即为sds的cmc 值。
[0174]
实施例11以化合物3为荧光指示剂测定ctab的cmc
[0175]
将化合物3配制成34μm的乙腈储备液,分别配制浓度为0.01、0.05、0.1、 0.2、0.3、0.4、0.5、0.6、0.7、0.8、0.9、1.0、1.2、1.5、2.0mm的ctab 水溶液。将探针储备液以1:200(v/v)比例加入至系列浓度的ctab溶液中,37℃下孵育4小时以上,待冷却至室温后,测定荧光。以最大激发、发射波长荧光值对ctab浓度作图,结果见附图20。荧光强度随ctab浓度的变化符合s型曲线,与化合物3在不同浓度ctab溶液中的包载状态一致。对s型曲线分别做切线,得到的第一个交点对应的浓度为0.80mm,即为ctab的cmc值。
[0176]
实施例12以化合物3为荧光指示剂测定mpeg
5k-pdlla
5k
的cmc
[0177]
将化合物3配制成3.4μm的乙腈储备液,分别配制浓度为0.001、0.005、 0.01、0.02、0.05、0.1、0.2、0.4、0.6、0.8、1.0、2.0、4.0、10.0μm的 mpeg
5k-pdlla
5k
水溶液。将探针储备液以1:200(v/v)比例加入至各溶液中待冷却至室温后,测定荧光。得到的化合物3荧光强度随mpeg
5k-pdlla
5k
浓度变化曲线见附图21,当mpeg
5k-pdlla
5k
浓度小于0.1μm时,化合物2因其在水溶液中聚集而完全无信号。随着mpeg
5k-pdlla
5k
浓度逐渐增加至0.1μm及以上时,化合物3荧光开始出现且逐渐增强。各数据点的分布同样极其符合s型曲线。对s 型曲线分别做切线,得到的第一个交点对应的浓度为0.11μm,即为 mpeg
5k-pdlla
5k
的cmc值。
[0178]
实施例13以化合物4-57为荧光指示剂测定sds、ctab、曲拉通x-100的 cmc
[0179]
以本发明氮杂-氟硼二吡咯甲川化合物4-57为荧光指示剂,采用本发明所述后孵育法、前孵育法、稀释法的步骤或其变化步骤,对阴离子表面活性剂sds、阳离子表面活性剂ctab、非离子表面活性剂曲拉通x-100的cmc进行测定,结果如表3所示。
[0180]
表3
[0181]
[0182][0183]
实施例14以化合物1、2、3为荧光指示剂测定多种表面活性剂的cmc
[0184]
以本发明氮杂-氟硼二吡咯甲川化合物1、2、3为荧光指示剂,采用本发明所述后孵育法、前孵育法、稀释法的步骤或其变化步骤,对其他常用表面活性剂吐温80、吐温20、聚氧乙烯氢化蓖麻油el、聚氧乙烯氢化蓖麻油elp、苄泽58、苄泽59、麦泽52的cmc进行测定,结果如表4所示。吐温80(a)、(b)分别由两个供应商提供,成分上可能存在一定的区别,因此cmc测定结果也有一定的差异。
[0185]
表4
[0186]
常用表面活性剂化合物1化合物2化合物3吐温80(a)(μm)2.772.752.78吐温80(b)(μm)2.512.522.56吐温20(μm)5.825.785.90聚氧乙烯氢化蓖麻油el(μm)1.521.451.53
聚氧乙烯氢化蓖麻油elp(μm)1.501.451.48苄泽58(μm)2.802.752.76苄泽59(μm)2.412.412.47麦泽52(μm)2.262.292.31
[0187]
。
[0188]
实施例15以化合物1、2、3为荧光指示剂测定多种两亲性嵌段聚合物的cmc
[0189]
以本发明氮杂-氟硼二吡咯甲川化合物1、2、3为荧光指示剂,采用本发明所述后孵育法、前孵育法、稀释法的步骤或其变化步骤,对代表性两亲性嵌段共聚物mpeg
5k-pdlla
3k
、mpeg
2.5k-pdlla
2.5k
、dspe-peg2000、poloxamer 407、普朗尼克p104、soluplus的cmc进行测定,结果如表5所示。
[0190]
[表5]
[0191] 化合物1化合物2化合物3mpeg
5k-pdlla
3k
(μm)0.230.220.22mpeg
2.5k-pdlla
2.5k
(μm)0.550.560.61dspe-peg2000(μm)0.280.260.31泊洛沙姆407(37℃)(μm)1.401.341.39普朗尼克p104(37℃)(μm)1.531.561.59soluplus(μm)0.0290.0220.027
[0192]
。
[0193]
实施例16以化合物1、2、3为荧光指示剂测定普朗尼克p104的临界胶束温度
[0194]
将化合物2配制成34μm的乙腈储备液,配制0.5mg/ml的普朗尼克p104 (p104)水溶液。将化合物2的储备液按照1:200(v/v)的比例加入至普朗尼克p104溶液中,依次放置于5、10、15、20、25、30、35及40℃的水浴中,通过水循环控制水浴温度(
±
0.5℃)。超声10min后,检测各样品的荧光。不同温度下样品中化合物2的荧光光谱见附图22。当温度低于p104的临界胶束温度 (cmt)时,p104处于单分子状态,难以包载化合物2,化合物2由于聚集而没有荧光;而当温度高于cmt时,p104逐渐形成胶束而包载p2,从而p2的荧光出现且迅速增加。以最大激发、发射波长测得的荧光值对温度的响应曲线见附图 23。p104溶液中的化合物2在温度升至25-30度时,荧光出现且迅速增加至最大值。因此,对于0.5mg/ml的p104,计算出的cmt(胶束刚开始形成的温度) 为27.7℃,符合文献报道的结果。
[0195]
实施例17,对比例1芘荧光法测定曲拉通x-100的cmc
[0196]
将芘配制成400μm的乙腈储备液,分别配制浓度为0.01、0.02、0.05、 0.1、0.2、0.25、0.3、0.4、0.5、0.6、0.8、1.0、1.5、2.0mm的曲拉通x-100 水溶液。取芘的储备液按1:200(v/v)比例加入至各浓度的曲拉通x-100溶液中,于37℃下避光、避氧震荡孵育12小时以上。待恢复至室温后,测定各样品的荧光。以335nm为固定激发波长,得到的各样品中芘荧光发射光谱见附图24。以372nm为固定发射波长,得到的各样品中芘荧光激发光谱见附图25。随着曲拉通x-100浓度的增加,芘探针的荧光逐渐增强且峰型发生略微改变。取发射光谱中第一个峰值(i
372
)与第三个峰值(i
384
)的比值(i1/i3)对曲拉通x-100浓度作图,如附图26所示。取曲线中心对应的浓度为曲拉通x-100的cmc值,即为0.25mm。取激发光谱中i
339
与i
333
的比值(i
339
/i
333
)对曲拉通x-100浓度作图,如附图25所示。对曲线做切线,得到的第一个交点
对应的浓度为0.23mm,即为曲拉通x-100的cmc值。
[0197]
将芘配制成400μm的乙腈储备液,分别配制浓度为0.0075、0.075、0.15、 0.375、0.75、1.5、3.75、7.5、11、15、30、75、150、300μm的吐温80水溶液。取芘的储备液按1:200(v/v)比例加入至各浓度的吐温80溶液中,于37℃下避光、避氧震荡孵育12小时以上。待恢复至室温后,测定各样品的荧光。以 335nm为固定激发,得到各样品中芘荧光发射光谱。以372nm为固定发射波长,得到各样品中芘荧光激发光谱。取发射光谱中第一个峰值(i
372
)与第三个峰值 (i
384
)的比值(i1/i3)对吐温80浓度作图,如附图27所示。取曲线中心对应的浓度为吐温80的cmc值,即为15.3μm。取激发光谱中i
339
与i
333
的比值 (i
339
/i
333
)对吐温80浓度作图,如附图27所示。对曲线做切线,得到的第一个交点对应的浓度为4.66μm,即为吐温80的cmc值。
[0198]
按上述方法连续配制3组样品,加入芘并孵育完全后,以372nm为固定发射波长,得到各样品中芘荧光激发光谱。绘制i
339
/i
333
对吐温80浓度图,如附图 28所示。各浓度下对应的3个比值变异大,对曲线所做的3条切线无法重合,得到的第一个交点变异大。经3组样品得到吐温80的cmc分别为6.17、5.45、 3.47mm,精密度为27.8%,精密度不佳。
[0199]
实施例18,对比例2以常用荧光染料为指示剂测定曲拉通x-100的cmc
[0200]
将hoechst 33342配制成400μm的甲醇储备液,分别配制浓度为0.01、 0.02、0.05、0.08、0.1、0.2、0.25、0.3、0.4、0.5、0.6、0.8、1、1.5、2mm 的曲拉通x-100水溶液。将hoechst 33342储备液按1:200(v/v)比例加入至系列浓度曲拉通x-100水溶液中,于37℃下避光孵育12小时以上。待完全冷却至室温后,测定各样品中hoechst 33342的荧光。以335nm为固定激发波长,得到各样品中hoechst 33342的荧光发射光谱,结果见附图29。无论样品中曲拉通x-100的浓度高于cmc抑或低于cmc,hoechst 33342一直具有较强的荧光。以最大发射波长处的荧光值对曲拉通x-100浓度作图,如附图30所示。虽然随着曲拉通x-100浓度的增加,探针逐渐被包载于胶束内部从而荧光逐渐增强。但是,在曲拉通x-100的cmc处,无法观察到明显的转折点。对突变区域趋势线作最大斜率切线,得到的第一个交点所对应的浓度0.25mm即为曲拉通x-100的 cmc值,明显大于采用本发明的氮杂-氟硼二吡咯甲川化合物测得的cmc值。
[0201]
实施例19,对比例3以aie荧光染料tpe为指示剂测定曲拉通x-100的 cmc
[0202]
将tpe配制成100μm的丙酮储备液,分别配制浓度为0.01、0.02、0.05、 0.1、0.2、0.25、0.3、0.4、0.5、0.6、0.8、1.0、2.0的曲拉通x-100溶液,按1:200(v/v)的比例加入tpe的丙酮溶液从而使每个样品中含有0.5μm tpe。超声20min后,室温下静置1h。随后,通过荧光分光光度计测定各样品荧光。以310nm为固定激发波长,得到的各样品中tpe荧光发射光谱见图31。由附图 31可知,当曲拉通x-100的浓度低于0.25mm时,tpe分子由于聚集,在400-500 nm处有强烈的荧光。而当曲拉通x-100浓度为0.25mm及以上时,tpe分子由于被包载,在400-500nm处的荧光迅速下降。取最大发射波长470nm处的荧光值对曲拉通x-100浓度作图,如附图32所示。取曲线中荧光不再下降的初始点作为曲拉通x-100的cmc,为0.25mm。值得注意是,该浓度为tpe被胶束完全包载时的曲拉通x-100浓度,因此相对于曲拉通x-100的真实cmc值偏高。
[0203]
配制10mm sds溶液,各溶液中含有10μm tpe,预先37℃孵育过夜后,待冷却至室温,分别使用去离子水稀释配制好的母液至浓度为10、9、8、7.5、 7、6.5、6、5、4、3mm,静置30
分钟后,以310nm为固定激发波长,测定各样品荧光值。以发射波长为450nm处的荧光值对sds浓度作图,如附图33所示。随着sds浓度的降低,tpe荧光总体上一直下降,并未出现文献报道的先上升后下降趋势,无法计算sds的cmc值。
[0204]
分别配制浓度为2、1、0.4、0.2、0.1、0.05、0.02mm的tpe丙酮储备液。随即将各储备液以1:200(v/v)比例加入至去离子水中,得到浓度分别为10、5、2、1、0.5、0.25、0.1μm的tpe水溶液,每个浓度同时配制3份。将配制好的tpe水溶液室温静置30分钟后,以310nm为固定激发波长,以450nm为固定发射波长,测定各样品的荧光强度。结果见附图34,水中聚集的tpe荧光强度与其浓度的线性较差,且荧光强度变异大。
[0205]
实施例,20,对比例4芘荧光指示剂用于测定两亲性嵌段共聚物 mpeg
5k-pdlla
5k
的cmc
[0206]
芘探针用于测定mpeg
5k-pdlla
5k
共聚物cmc的结果如图35所示,当共聚物浓度低于1μm时,i
339
/i
333
的比值在0.2-0.3间波动;当共聚物浓度大于1μm 时,i
339
/i
333
逐渐增加。由于i
339
/i
333
在本底区间(0.2-0.3)波动较大,致使转折点并不明显。并且,各批次间比值变异同样较大,通过切线法得到各批次测得的 cmc分别为1.18、0.90、0.39μm。
[0207]
实施例21,对比例5以对比化合物1为荧光指示剂测定曲拉通x-100的 cmc。
[0208]
对比化合物1分子结构式如下:
[0209][0210]
将对比化合物1配制成34μm的乙腈储备液,分别配制浓度为0.005、0.01、 0.025、0.04、0.05、0.1、0.125、0.15、0.2、0.25、0.3、0.4、0.5、0.75mm 的曲拉通x-100水溶液。将探针储备液以1:200(v/v)比例加入至系列浓度曲拉通x-100溶液中,37℃下孵育12小时以上,待冷却至室温后,测定荧光。以 537nm为固定激发波长,得到各样品中对比化合物1的荧光发射光谱,结果见附图36。无论样品中曲拉通x-100的浓度高于cmc抑或低于cmc,对比化合物1 一直具有较强的荧光。以最大发射波长处(571nm)的荧光值对曲拉通x-100 浓度作图,结果如图37所示,随着曲拉通x-100浓度的增加,探针的荧光信号逐渐下降,无法观察到明显的转折点。无法对cmc值进行准确计算。
[0211]
实施例22,对比例6以对比化合物2为荧光指示剂测定曲拉通x-100的cmc 对比化合物2分子结构式如下:
[0212][0213]
将化合物2配制成34μm的乙腈储备液,分别配制浓度为0.005、0.01、 0.025、0.04、
0.05、0.1、0.125、0.15、0.2、0.25、0.3、0.4、0.5、0.75mm 的曲拉通x-100水溶液。将探针储备液以1:200(v/v)比例加入至各曲拉通x-100 溶液中,37℃下孵育12小时以上,待冷却至室温后,测定荧光。以564nm为固定激发波长,得到各样品中对比化合物2的荧光发射光谱,结果如图38所示。无论样品中曲拉通x-100的浓度高于cmc抑或低于cmc,对比化合物2一直具有荧光。以最大发射波长处(610nm)的荧光值对曲拉通x-100浓度作图,结果见如图39所示,当曲拉通x-100浓度低于cmc时,对比化合物2的荧光较弱,但随着曲拉通浓度增加,对比化合物2的荧光逐渐增强;当曲拉通x-100浓度增加至其cmc以上时,对比化合物2的荧光增强更为明显。由于对比化合物2在曲拉通x-100浓度高于或低于cmc时皆具有荧光,荧光变化的转折点并不明显。并且,在曲拉通x-100浓度低于cmc时对比化合物2的荧光已经呈现逐渐增强的趋势,这极容易误导研究者认为此时曲拉通x-100已经开始形成胶束。因此,通过切线法得到的cmc值为0.156mm,结果明显小于文献报道的0.20mm。
[0214]
实施例23,对比例7以对比化合物2为荧光指示剂测定sds的cmc
[0215]
将化合物2配制成34μm的乙腈储备液,分别配制浓度为0.1、0.5、1、3、 4、5、6、7、8、9、10、15、20mm的sds水溶液。将探针储备液以1:200(v/v) 比例加入至各sds溶液中,37℃下孵育12小时以上,待冷却至室温后,测定荧光。以564nm为固定激发波长,得到各样品中对比化合物2的荧光发射光谱,结果见附图40。由图可知,无论sds浓度高于或低于其cmc,化合物2始终具有较强的荧光信号。以最大发射波长(610nm)的荧光值对sds浓度作图,结果见如图41所示,荧光强度随sds浓度的变化呈单调增强的趋势,图中无法观察到明显的突变点,并且无法通过切线法对sds的cmc值进行计算。
[0216]
实施例24,对比例8以对比化合物3为荧光指示剂测定曲拉通x-100的cmc 对比化合物3分子结构如下:
[0217][0218]
将对比化合物3配制成34μm的乙腈储备液,分别配制浓度为0.005、0.01、 0.025、0.04、0.05、0.1、0.125、0.15、0.2、0.25、0.3、0.4、0.5、0.75mm 的曲拉通x-100水溶液。将探针储备液以1:200(v/v)比例加入至系列浓度的曲拉通x-100溶液中,37℃下孵育12小时,待冷却至室温后,测定荧光。以562 nm为固定激发波长,得到各样品中化合物2的荧光发射光谱,结果如图42所示。无论样品中曲拉通x-100的浓度高于cmc抑或低于cmc,对比化合物3一直具有荧光。以最大发射波长处(608nm)的荧光值对曲拉通x-100浓度做曲线,结果见附图43。当曲拉通x-100浓度低于cmc时,对比化合物3的荧光已经出现且逐渐增强;当曲拉通x-100浓度高于其cmc时,对比化合物3的荧光增强更为明显。由于对比化合物3在曲拉通x-100浓度高于或低于cmc时皆具有荧光,荧光变化的转折点并不明显,并且,在曲拉通x-100浓度低于cmc时对比化合物3 的荧光已经呈现逐渐增强的趋势,这极容易被误认为此时曲拉通x-100已经开始形成胶束。因此,通过切线法得到的cmc值为0.153mm,结果明显小于文献报道的0.20mm。
[0219]
实施例25,对比例9以对比化合物3为荧光指示剂测定sds的cmc
[0220]
将对比化合物3配制成34μm的乙腈储备液,分别配制浓度为0.1、0.5、 1、3、4、5、6、7、8、9、10、15、20mm的sds水溶液。将探针储备液以1:200 (v/v)比例加入至各sds溶液中,37℃下孵育12小时以上,待冷却至室温后,测定荧光。以562nm为固定激发波长,得到各样品中对比化合物3的荧光发射光谱,结果见附图44。由图可知,无论sds浓度高于或低于其cmc,对比化合物 3始终具有较强的荧光信号。以最大发射波长(608nm)的荧光值对sds浓度作图,结果如图45。荧光强度随sds浓度的变化呈单调增强的趋势。与其他氟硼二吡咯甲川化合物相似,同样无法从图中观察到明显的突变点,并且无法通过切线法对sds的cmc值进行计算。
[0221]
实施例26,对比例10以dph为荧光指示剂测定dspe-peg2000的cmc
[0222]
将dph配制成50μm的四氢呋喃储备液,分别配制浓度为0.04、0.02、0.07、 0.18、0.36、0.71、1.4、2.1、2.9、3.6、7.1、14.3、35.7μm的dspe-peg2000 水溶液。将dph储备液按1:200(v/v)比例加入至系列浓度的dspe-peg2000水溶液中,37℃下孵育12小时。待完全冷却至室温后,测定各样品中dph的荧光。以355nm为固定激发波长,得到各样品中dph的荧光发射光谱,结果见附图46。无论样品中dspe-peg2000的浓度高于cmc抑或低于cmc,dph一直具有较强的荧光。以最大发射波长处的荧光值对dspe-peg2000浓度作图,如图47所示。当 dspe-peg2000浓度低于其cmc时,dph的荧光具有较强的波动性,而进一步随着 dspe-peg2000浓度的增加至其cmc以上时,探针逐渐被包载于胶束内部从而荧光逐渐增强。但是,无法获得明显的s型曲线。通过切线法得到的第一个交点对应的浓度为1.1μm,大于其他方法得到的cmc值。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。