1.本发明涉及物信息分析技术领域,具体地说,涉及一种利用单细胞测序数据计算池测序中细胞亚群富集分数的方法。
背景技术:
2.肿瘤是一种包含多种细胞种类的病理组织。包括但不限于肿瘤细胞,免疫细胞,基质细胞,内皮细胞等。细胞组成的比例差异决定了肿瘤的恶性程度,转移能力,对治疗的反应性等。
3.单细胞转录组技术是结合细胞分离技术与二代测序技术的新兴技术手段。其主要特点为可获得单个细胞的基因表达谱,可在基因表达层面对细胞亚群分群,可以对不同的细胞群及其基因表达的差异,生理功能差异进行研究。可用于稀有细胞群的发现,鉴定。此技术尤其适合解析复杂细胞群,例如干细胞,胚胎细胞,肿瘤细胞的细胞种类构成。
4.池测序为传统的二代测序技术,针对细胞群进行测序,获得较为笼统,粗糙的细胞群的表达信息。其中非主要细胞群的表达信息往往被掩盖,很多重要信息,尤其是细胞构成的比例信息无法获得。
5.池测序由于发展时间长,成本低,对计算资源要求小的优势,有充足的数据资源可供获取,分析。目前较为知名的,肿瘤相关的池测序数据集包括tcga,icgc,geo数据库,包含了成百上千例临床随访信息完善的池测序样本,为肿瘤研究提供了丰富的资源。
6.单细胞转录组测序虽然有分辨率高,可以解析样本内细胞组成等优势,但由于其成本高,发展时间较短,目前尚无包含临床信息的单细胞转录组测序的数据集。研究一种将单细胞测序对细胞组成的解析结果用于池测序,推测出池测序内细胞组成的富集分数的方法,从而结合两种测序的优势,对肿瘤研究有重要意义。
7.专利文献cn112700820a公开了一种基于单细胞转录组测序的细胞亚群注释方法,包括如下步骤:1)10x barcode umi识别,2)比对基因组,3)基因表达谱,4)低质量细胞过滤和数据均一化,5)细胞群体聚类,6)marker基因提取,7)细胞亚群注释。该发明解决了单细胞亚群注释的问题,使得单细胞测序数据在常规分析后,可以支持依据基因表达谱和/或细胞marker基因进行细胞注释,实现了不同注释方法的有机结合,得到细胞类型的分布情况和相关信息。然而该文献涉及的是单细胞测序结果细胞群的注释,目前未见如本技术的利用单细胞测序数据来计算池测序中细胞亚群富集分数的方法。
技术实现要素:
8.本发明的目的是针对现有技术中的不足,提供一种利用单细胞测序数据计算池测序中细胞亚群富集分数的方法。
9.本发明的再一的目的是,提供一种利用单细胞测序数据计算池测序中细胞亚群富集分数的系统。
10.为实现上述第一个目的,本发明采取的技术方案是:
11.一种利用单细胞测序数据计算池测序中细胞亚群富集分数的方法,包括以下步骤:
12.a)通过单细胞测序,建立marker基因名,细胞亚群名的表达矩阵m;
13.b)通过池测序,建立基因名,样本名的表达矩阵n;
14.c)将所述marker基因名,细胞亚群名的表达矩阵m中的marker基因名比对至所述基因名,样本名的表达矩阵n,获取marker基因名,样本名的表达矩阵n’;
15.d)使用程序scfrac,以m,n’为输入,得到m中每个细胞亚群在n’的各个样本中所占的富集分数。
16.作为本发明的一个优选例,步骤a所述的单细胞测序为单细胞转录组测序。
17.更优选地,所述单细胞转录组测序的方法选自smart-seq、smart-seq2、cel-seq、cel-seq2、drop-seq、mars-seq、mars-seq2和scrb-seq。
18.作为本发明的另一优选例,步骤a所述的单细胞测序采用的单细胞分离和标记平台为chromium
tm
系统、bd rhapsody
tm
单细胞分析系统、单细胞测序解决方案、icell8单细胞系统或c1
tm
单细胞全自动制备系统。
19.作为本发明的另一优选例,步骤a所述的单细胞测序采用的单细胞高通量测序平台为illumina系列、bgiseq系列、roche 454、abi solid或ion proton。
20.作为本发明的另一优选例,步骤a包括以下步骤:
21.a-1)10xbarcode umi识别:10x genomics平台建库测序的下机数据为fastq序列,包括barcode,umi,mrna序列三部分,使用软件cellranger count,通过barcode序列识别细胞,通过umi序列对基因表达定量,通过3’端mrna序列进行基因鉴定;
22.a-2)比对基因组:采取star算法,将fastq序列比对至参考基因组上,获得序列的基因信息,使用cellranger对基因的表达量进行定量;
23.a-3)低质量细胞过滤和均一化:基于细胞的基因表达量和线粒体dna表达量对低质量细胞过滤,使用r语言的seurat包,过滤细胞之后,再通过seurat包的normalization函数对表达量进行均一化;
24.a-4)细胞聚类:使用主成分分析方法对细胞表达谱降维,选取前10个主成分用于后续的聚类,使用临近算法对细胞进行聚类,使用umap对细胞进行基于图论的可视化聚类,将knn获得的聚类结果映射至umap聚类结果上;
25.a-5)marker基因提取:seurat通过wilcox法比较检验不同细胞组成间的差异基因,其中表达较其他细胞群体显著上升的基因即为该细胞亚群的marker基因;
26.a-6)建立marker基因,目标细胞群表达矩阵m:将每个细胞亚群的marker基因作为行名,每个单细胞作为列名,从单细胞测序数据中获得一表达矩阵m。
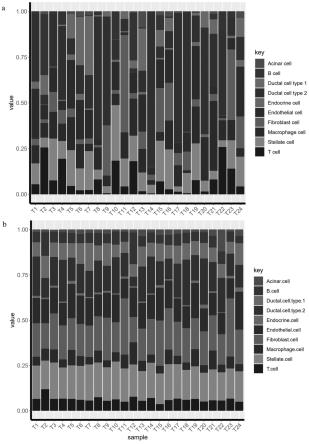
27.作为本发明的另一优选例,步骤b包括以下步骤:从组织提取全组织rna,经过反转录为cdna,片段化为cdna片段,两端加引物,pcr扩增,测序,从illumina平台测序下机的数据包括fastq_1,fastq_2,经过fastq质控,star比对至参考基因组,htseq-count获取基因表达量,获取以基因为行名,样本为列名的池测序的表达矩阵n。
28.为实现上述第二个目的,本发明采取的技术方案是:
29.一种利用单细胞测序数据计算池测序中细胞亚群富集分数的系统,所述系统用于
将单细胞测序建立的marker基因名,细胞亚群名的表达矩阵m中的marker基因名比对至池测序建立的基因名,样本名的表达矩阵n,进而获取marker基因名,样本名的表达矩阵n’,最终获得m中每个细胞亚群在n’的各个样本中所占的富集分数。
30.本发明优点在于:
31.1、本发明提供了一种分析样本内部各细胞亚群所占比例的方法。大型临床队列的样本包含有丰富的生物信息,但单细胞测序价格昂贵,对取材要求高,导致样本例数纳入少,从而产生数据偏倚,也无法用单细胞测序技术探究其表达谱特征与细胞组分。而本发明可以利用池测序(如二代测序)价格低廉,标本处理要求低,技术成熟的优势,有机结合单细胞测序,能够更好的分析样本内部各细胞的生物学特征及比例,也可在池测序队列中验证和进一步探究单细胞测序的结果。尤其适合在大样本队列中寻找肿瘤内部的细胞亚群与临床意义之间的关联性。本发明的技术有助于减少前期的实验投入,节省科研经费的开支,有的放矢。
32.2、本发明利用机器学习的方式拟合得到一个包括单细胞测序和池测序的模型,该模型用于推断池测序中细胞比例具有准确度高的优势。
附图说明
33.附图1是实施例1的单细胞测序中的细胞聚类分析结果。
34.附图2是实施例1中单细胞测序所得的细胞亚群比例(a)与按本发明方法推测的细胞亚群比例(b)的比较。
35.附图3是实施例1中b细胞(a),fibroblast细胞(b)经单细胞测序所得富集分数和按本发明方法推测的富集分数的相关性分析。
具体实施方式
36.下面结合附图对本发明提供的具体实施方式作详细说明。
37.实施例1在胰腺癌中利用单细胞测序数据计算池测序中细胞亚群富集分数
38.0、取15份新鲜的胰腺癌组织,平均分为两份,一份进行10x genomic单细胞测序,另外一份行rnaseq池测序。
39.1、10xbarcode umi识别:10x genomics平台建库测序的下机数据为fastq序列,包括barcode,umi,mrna序列三部分。使用软件cellranger count,通过barcode序列识别细胞,通过umi序列对基因表达定量,通过3’端mrna序列进行基因鉴定。
40.2、比对基因组:采取star算法,将fastq序列比对至参考基因组上,获得序列的基因信息。使用cellranger对基因的表达量进行定量。
41.3、低质量细胞过滤和均一化:基于细胞的基因表达量和线粒体dna表达量对低质量细胞过滤。使用r语言的seurat包,过滤细胞之后,再通过seurat包的normalization函数对表达量进行均一化。
42.4、细胞聚类:使用pca(主成分分析)方法对细胞表达谱降维,选取前10个主成分用于后续的聚类。使用knn(临近算法)对细胞进行聚类,使用umap对细胞进行基于图论的可视化聚类。将knn获得的聚类结果映射至umap聚类结果上。(图1)
43.5、marker基因提取:seurat通过wilcox法比较检验不同细胞组成间的差异基因,
其中表达较其他细胞群体显著上升的基因即为该细胞亚群的marker基因。
44.6、建立marker基因,目标细胞群表达矩阵m:将每个细胞亚群的marker基因作为行名,每个单细胞作为列名,从单细胞测序数据中获得一表达矩阵m。
45.7、池测序:从组织提取全组织rna,经过反转录为cdna,片段化为cdna片段,两端加引物,pcr扩增,测序。从illumina平台测序下机的数据包括fastq_1,fastq_2。经过fastq质控,star比对至参考基因组,htseq-count获取基因表达量,获取以基因为行名,样本为列名的池测序的表达矩阵n。
46.8、获取表达谱:将6中的marker基因比对至7中的表达矩阵n,获取以marker基因为行名,样本名为列名的表达谱n’。
47.9、使用程序scfrac获取细胞亚群的富集分数:使用程序scfrac,以m,n’为输入,得到m中每个细胞亚群在n’中所占的富集分数。
48.10、以条形图比较单细胞测序所得的细胞亚群比例(图2a)与经scfrac推测的细胞亚群比例(图2b),可见各个细胞比例较为接近。比较b细胞(图3a),fibroblast细胞(图3b)在单细胞测序和池测序中经scfrac推测的富集分数的相关性,可见相关性较强。
49.以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员,在不脱离本发明方法的前提下,还可以做出若干改进和补充,这些改进和补充也应视为本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。