1.本发明属于有机合成技术领域,具体涉及基于萘二酰亚胺的共轭大环材料及其制备方法。
背景技术:
2.共轭分子可简单描述为单双键交替相连的碳链分子,而如果单双键交替相连的碳链首位相连则构成了共轭大环分子。有机共轭分子是有机光电材料的重要组成部分,相比于无机光电材料,具有价廉、柔性、质量轻等优点。有机共轭分子主要有聚合物和小分子两大类,而共轭大环分子作为一类典型的小分子,相比于聚合物结晶性更好,可制备单晶器件。因此有机共轭大环分子的合成具有重要意义。萘酰亚胺类分子是一类典型的n型半导体分子,通常用作高迁移率的有机半导体材料。
3.另一方面,已经报道有在环状共轭分子的空腔内可以与一些客体分子自组装,如2018年报道的共轭环结构在其环空腔中可选择性结合碘离子(fully conjugated[4]chrysaorene.redox-coupled anion binding in a tetraradicaloid macrocycle,j.am.chem.soc.2018,140,14474-14480),也有与c
60
、c
70
等客体自组装的其他大环分子被相继被报道。并且由于环内空腔大小的可调控性,方便进一步研究具有不同大小的空腔的共轭分子与不同客体之间的选择性结合作用,在气体分离等领域有广泛应用前景。
技术实现要素:
[0004]
本发明的目的在于提供一种基于萘二酰亚胺的共轭大环材料及其制备方法。本发明将萘二酰亚胺结构引入大环体系,分别与菲单元和苯单元通过suzuki偶联合成共轭大环分子,为有机半导体材料提供了新的思路。
[0005]
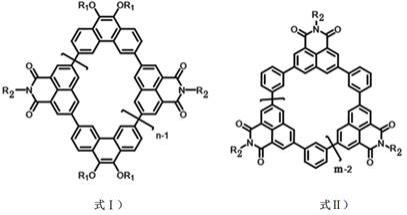
一类基于萘二酰亚胺的共轭大环材料,其结构通式分别如式ⅰ)或式ⅱ)所示:
[0006][0007]
式中,r1选自直链或支链烷基,r2选自芳烃、直链或支链烷基,n、m表示基本组成单元数,n为2、3或4,m为3、4或5。
[0008]
优选的,r1取代基为c
1-c
10
直链或支链烷基,r2选自c
1-c
10
直链或支链烷基、或以下
取代芳基中的一种:
[0009][0010]
其中,r3、r4、r5取代基为c
1-c
10
直链或支链烷基。
[0011]
本发明进一步提供一种上述的基于萘二酰亚胺的共轭大环材料的制备方法,以溴代物、硼酸酯为起始原料,经suzuki偶联反应,分别制得式ⅰ)、式ⅱ)所示的共轭大环材料分子;其中:
[0012][0013]
当r2为直链或支链烷基时,式ⅰ)、式ⅱ)所示的共轭大环材料分子的合成路线如下:
[0014][0015]
本发明提供的基于萘二酰亚胺的共轭大环材料的制备方法,包括以下步骤:在有机溶剂和水中,碱、催化剂及配体存在的情况下,以溴代物a与硼酸酯b,或者以硼酸酯a1与溴代物b1为起始原料,进行suzuki偶联反应,通过hplc分离得到式ⅰ)所示化合物,以溴代物c与硼酸酯b,或者以硼酸酯c1与溴代物b1为起始原料,进行suzuki偶联反应,通过hplc分离得到式ⅱ)所示化合物。
[0016]
优选的,有机溶剂选自苯、甲苯、二甲苯、氯代苯、十氢化萘、乙醚、二乙醚、二异丙基醚、甲基叔丁基醚、甲基叔戌基醚、苯甲醚、二恶烷、四氢呋喃、2-甲基四氢呋喃、1,2-二甲氧基乙烷、1,2-二乙氧基乙烷和1,1-二乙氧基甲烷中的一种;有机溶剂和和水的体积比为1:2-2:1。
[0017]
优选的,所述的有机溶剂与硼酸酯的体积物质的量比优选为300ml/mmol~600ml/mmol。
[0018]
优选的,碱选自叔丁醇钠、叔丁醇钾、碳酸钾、碳酸钠和碳酸铯中的一种;催化剂为双二亚苄基丙酮钯pd(dba)2、三(二亚苄基丙酮)二钯pd2(dba)3、四(三苯基膦)钯pd(pph3)4、二氯化钯、[1,1'-双(二苯基膦)二茂铁]二氯化钯pd(dppf)cl2、醋酸钯和氯(2-二环己基膦基-2',4',6'-三异丙基-1,1'-联苯基)[2-(2'-氨基-1,1'-联苯)]钯(ii)xphospd g2的一种;配体为三苯基膦、三叔丁基膦、三叔丁基磷四氟硼酸盐[(t-bu
)3
ph]bf4)、1,2,3,4,5-戊苯基-1'-(二叔丁基磷基)二茂铁qphos、2-二环己基膦-2'-甲基联苯mephos和2-二环己基膦-2,4,6-三异丙基联苯xphos中的一种。
[0019]
优选的,硼酸酯和溴代物的摩尔比值均为1.0,配体和催化剂的摩尔比为0:1~5:1。
[0020]
优选的,反应温度为25℃-80℃。
[0021]
进一步优选的,基于萘二酰亚胺的共轭大环材料的制备方法包括如下步骤:
[0022]
1)在反应容器中加入硼酸酯、溴代物、氯(2-二环己基膦基-2',4',6'-三异丙基-1,1'-联苯基)[2-(2'-氨基-1,1'-联苯)]钯(ii),2mol/l磷酸钾水溶液,惰性气体氛围下,
加入四氢呋喃,保持温度为25℃-80℃,持续搅拌反应16-32小时;其中:硼酸酯和溴代物的摩尔比为1:1,氯(2-二环己基膦基-2',4',6'-三异丙基-1,1'-联苯基)[2-(2'-氨基-1,1'-联苯)]钯(ii)和硼酸酯的摩尔比为1:10,四氢呋喃和体系中水的体积比为1:2-2:1;
[0023]
2)反应达到预定时间后,采取tlc、hplc或nmr监测,硼酸酯或溴代物消失后停止反应,减压蒸馏除去体系中的四氢呋喃,用三氯甲烷萃取,有机相经分离纯化即得到大环产物。
[0024]
进一步优选的,硼酸酯b通过以下方法制备得到:
[0025]
(1)以1,8-萘二酸酐为起始原料,以浓硫酸为溶剂,在nbs溴化剂作用下,在55-65℃的温度下发生溴代反应,制得
[0026]
(2)以伯胺化合物r2nh2为原料,制得即溴代物b1;
[0027]
(3)以溴代物b1和联硼酸频那醇酯为原料,以pdcl2为催化剂,在醋酸钾作用下,在溶剂中加热反应制得硼酸酯b。
[0028]
进一步优选的,硼酸酯a1通过以下方法制备得到:
[0029]
(1)在氮气氛围下,称取3,6-二溴菲9,10-二酮、连二亚硫酸钠和四丁基溴化铵溶于四氢呋喃和水的混合溶剂中,先反应20-40分钟,然后加入碳酸钾水溶液、溴化物r1br,加热回流反应,制得即溴代物a;
[0030]
(2)以溴代物a和联硼酸频那醇酯为原料,以pdcl2为催化剂,在醋酸钾作用下,在溶剂中加热反应制得硼酸酯a1。
[0031]
与现有的技术相比,本发明所述的基于萘二酰亚胺的共轭大环材料具有以下特点:
[0032]
1)提供了一种自下而上合成小分子共轭大环材料的合成路线,其合成路线简单有效,可重复性好;在单体合成中,使用全新方法只需一步合成烷基取代的二溴萘酰亚胺中间产物(详见实施例4,大大简化了原有的合成路线。
[0033]
2)本发明设计合成的一系列具有不同环内空腔的大环分子,因其每个分子中都存在大小不同的环内纳米孔,根据环内纳米孔大小不同,可以选择性地结合不同的客体分子进行超分子组装,在气体分离等有良好的应用。
附图说明
[0034]
图1:化合物7的核磁氢谱(在氘代氯仿中),表征实施例7中的化合物7。
[0035]
图2:化合物8的核磁氢谱(在氘代氯仿中),表征实施例7中的化合物8。
[0036]
图3:化合物9的核磁氢谱(在氘代氯仿中),表征实施例8中的化合物9。
[0037]
图4:化合物10的核磁氢谱(在氘代氯仿中),表征实施例8中的化合物10。
[0038]
图5:化合物11的核磁氢谱(在氘代氯仿中),表征实施例9中的化合物11。
[0039]
图6:化合物12的核磁氢谱(在氘代氯仿中),表征实施例9中的化合物12。
[0040]
图7:化合物7的单晶结构图。
[0041]
图8:化合物11与c70核磁滴定。
[0042]
图9:化合物12与c70核磁滴定。
[0043]
图10:化合物11与c70荧光滴定。
[0044]
图11:化合物12与c70荧光滴定。
[0045]
图12:化合物11根据荧光滴定拟合的结合常数。
[0046]
图13:化合物12根据荧光滴定拟合的结合常数。
具体实施方式
[0047]
下面通过实施例的方式进一步说明本发明,但并不因此将本发明限制在所述的实施例范围之中。下列实施例中未注明具体条件的实验方法,按照常规方法和条件,或按照商品说明书选择。
[0048]
实施例1:化合物1的制备
[0049][0050]
称取1,8-萘二酸酐(19.8g,99.9mmol)、n-溴代琥珀酰亚胺(nbs;40.9g,230mmol),加入浓硫酸(100ml),加热至60℃,反应6小时。反应结束后,冷却至室温,将混合物缓慢加入冰水(200ml)中,过滤收集沉淀物,在水(200ml)和乙腈(200ml)中洗涤,真空下干燥。加入n,n-二甲基甲酰胺(dmf;250ml),加热至完全溶解,静止12小时,过滤沉淀真空下干燥后得到11g粉红色固体1,产率34%。
[0051]
实施例2:化合物2的制备
[0052][0053]
称取化合物1(7.12g,20mmol)、邻二异丙基苯胺(3.54g,60mmol),加入醋酸(80ml),加热至120℃,反应12小时。反应结束后,冷却至室温,将混合物缓慢加入冰水(100ml)中,过滤收集沉淀物,在乙腈(100ml)中洗涤,粗产物经过经硅胶柱层析(二氯甲烷:石油醚=1:1)分离提纯,真空下干燥后得到7.6g白色固体2,产率74%。
[0054]
实施例3:化合物3的制备
[0055][0056]
在氮气氛围下,称取化合物2(4g,7.75mmol)、联硼酸频那醇酯(5.9g,23.25mmol)、醋酸钾(3.8g,38.7mmol)、pdcl2(dppf)
·
ch2cl2(100mg,0.12mmol),加入干燥二氧六环(80ml),加热至80℃,反应12小时。反应结束后,除去体系中的二氧六环,粗产物经过经硅胶柱层析(洗脱液:二氯甲烷)分离后,用甲醇(50ml)洗涤,真空下干燥后得到3.9g白色固体3,产率84%。
[0057]
实施例4:化合物4的制备
[0058][0059]
称取化合物1(3.56g,10mmol)、正丁胺(730mg,10mmol),加入干燥二氯甲烷(dcm;50ml),加热至70℃,回流2小时后除去体系中的二氯甲烷;加入二氯亚砜(50ml),加热至70℃,反应6小时。反应结束后,除去体系中的二氯亚砜,粗产物经过经硅胶柱层析(二氯甲烷:石油醚=3:2)分离后,真空下干燥后得到1.2g白色固体4,产率29%。
[0060]
实施例5:化合物5的制备
[0061][0062]
在氮气氛围下,称取3,6-二溴菲9,10-二酮(3.66g,10mmol)、连二亚硫酸钠(10.44g,60mmol)、四丁基溴化铵(1.61g,0.05mmol),溶于600ml溶剂(四氢呋喃:水=1:1)中,反应30分钟。然后加入100ml碳酸钾(4g,29mmol)水溶液、溴丁烷(3.43g,25mmol),加热回流至110℃,反应48小时。反应结束后,除去体系中的水和四氢呋喃,粗产物经硅胶柱层析分离(石油醚:二氯甲烷=10:1)后,真空下干燥后得到3.5g
[0063]
白色固体5(3,6-二溴-9,10-二丁氧基菲),产率73%。
[0064]
实施例6:化合物6的制备
[0065][0066]
在氮气氛围下,称取化合物5(4.8g,10mmol)、联硼酸频那醇酯(7.6g,30mmol)、醋酸钾(4.9g,50mmol)、pdcl2(dppf)
·
ch2cl2(100mg,0.12mmol),加入干燥二氧六环(100ml),加热至80℃,反应12小时。反应结束后,除去体系中的二氧六环,粗产物经过经硅胶柱层析(二氯甲烷:石油醚=1:1)分离后,真空下干燥后得到3.1g白色固体6,产率54%。
[0067]
实施例7:化合物7、8的制备
[0068][0069]
在氮气氛围下,称取化合物3(280mg,0.46mmol)、化合物5(221mg,0.46mmol)、xphospd g2(40mg,0.05mmol),加入125ml四氢呋喃和250ml磷酸钾水溶液(2mol/l),加热至50℃,反应24小时。反应结束后,除去反应体系中的水和四氢呋喃,粗产物经硅胶柱层析分离(二氯甲烷:石油醚:乙酸乙酯=10:5:1)提纯后,通过hplc分离提纯,真空下干燥得到化合物7为25mg,产率8%;得到化合物8为15mg,产率3%。我们通过在氯仿中缓慢挥发溶剂获得了化合物7的单晶。化合物7的核磁氢谱,单晶结构分别见图1和图7;化合物8的核磁氢谱图见图2。化合物7:1h nmr(400mhz,cdcl3)δ9.39(s,4h),9.18(s,8h),8.48(d,j=8.5hz,4h),8.34(d,j=8.5hz,4h),7.52(d,j=7.8hz,2h),7.38(d,j=7.8hz,8h),4.34(t,j=6.6hz,8h),2.92
–
2.77(m,4h),2.05
–
1.92(m,8h),1.67(dd,j=15.1,7.5hz,8h),1.21(d,j=6.8hz,24h),1.07(t,j=7.4hz,12h).化合物8:1h nmr(400mhz,cdcl3)δ8.95(d,j=1.5hz,6h),8.80(s,6h),8.54(s,6h),8.45(d,j=8.5hz,6h),7.97(d,j=8.5hz,6h),7.45(t,j=7.7hz,3h),7.27
–
7.23(m,6h),4.36(t,j=6.6hz,12h),2.74(dt,j=13.5,6.7hz,6h),2.04
–
1.97(m,12h),1.68(dt,j=14.8,7.4hz,12h),1.14
–
1.08(m,18h),1.04(d,j=6.8hz,36h).
[0070]
实施例8:化合物9、10的制备
[0071]
[0072]
在氮气氛围下,称取化合物4(205mg,0.5mmol)、化合物6(287mg,0.5mmol)、xphospd g2(40mg,0.05mmol),加入125ml四氢呋喃和250ml磷酸钾水溶液(2mol/l),加热至50℃,反应24小时。反应结束后,除去反应体系中的水和四氢呋喃,粗产物经硅胶柱层析分离(二氯甲烷:石油醚:乙酸乙酯=10:5:1)提纯后,通过hplc分离提纯,真空下干燥得到化合物9为7mg,产率2.5%;得到化合物10为10mg,产率3.5%。化合物9的核磁氢谱及化合物10的核磁氢谱图见图3和图4。化合物9:1h nmr(400mhz,cdcl3)δ8.99(s,4h),8.78(s,4h),8.64(s,4h),8.21(s,4h),7.97(s,4h),4.28(s,8h),4.22(s,4h),2.03
–
1.93(m,8h),1.77(d,j=8.2hz,4h),1.73
–
1.66(m,8h),1.52(d,j=7.3hz,4h),1.13(t,j=7.3hz,12h),1.04(t,j=7.3hz,6h).化合物10:1h nmr(400mhz,cdcl3)δ8.84(s,6h),8.68(s,6h),8.39(d,j=8.6hz,6h),8.37(s,6h),7.88(d,j=8.4hz,6h),4.31(t,j=6.6hz,12h),4.15(s,6h),1.95(dd,j=14.6,7.1hz,12h),1.70
–
1.64(m,6h),1.63(s,12h),1.36(d,j=7.8hz,6h),1.07(d,j=7.4hz,18h),0.82(t,j=7.2hz,9h).
[0073]
实施例9:化合物11、12的制备
[0074][0075]
在氮气氛围下,称取化合物3(304mg,0.5mmol)、1,3-二溴苯(118mg,0.5mmol)、xphospd g2(40mg,0.05mmol),加入125ml四氢呋喃和250ml磷酸钾水溶液(2mol/l),加热至50℃,反应24小时。反应结束后,除去反应体系中的水和四氢呋喃,粗产物经硅胶柱层析分离(二氯甲烷:石油醚:乙酸乙酯=10:5:1)提纯后,通过hplc分离提纯,真空下干燥得到化合物11为7mg,产率2.5%;得到化合物10为10mg,产率3.5%。化合物11的核磁氢谱及化合物12的核磁氢谱图见图5和图6。化合物11氢谱:1h nmr(400mhz,cdcl3)δ8.96(s,6h),8.48(s,6h),8.01(s,3h),7.88(d,j=7.8hz,6h),7.74(t,j=7.7hz,3h),7.51(t,j=7.7hz,3h),7.37(d,j=7.8hz,7h),2.82(dt,j=13.6,6.7hz,6h),1.18(dd,j=19.2,6.8hz,36h).化合物12氢谱:1h nmr(400mhz,cdcl3)δ9.03(s,8h),8.69(s,8h),8.30(s,4h),7.97(s,8h),7.76(s,4h),7.49(s,4h),7.32(d,j=7.7hz,8h),2.77(d,j=6.5hz,8h),1.14(d,j=6.8hz,48h).
[0076]
实施例10:化合物11,化合物12与富勒烯c70的超分子组装作用
[0077]
为了研究大环分子与c70的超分子组装作用,我们分别将化合物11与化合物12与富勒烯c70进行核磁滴定,如图8(化合物11与c70的核磁滴定),图9(化合物12与c70的核磁滴定)所示,将c70逐次滴入时,大环方向区的质子峰均发生了明显的移动,这说明大环分子与富勒烯存在有π-π相互作用,我们为了量化大环与富勒烯c70的结合力大小,我们又对他们进行了荧光滴定,如图10(化合物11与c70的荧光滴定),图11(化合物12与c70的荧光滴
定)所示,荧光滴定结果显示,c70的加入对大环的荧光有淬灭效果,这进一步证实了我们的想法,我们利用荧光滴定的结果,取荧光滴定图中荧光最强的点(512nm处),作出c70滴加当量与最强荧光的相关图,如图12(c70滴加当量与化合物11最强荧光的关系图),图13所示(c70滴加当量与化合物12最强荧光的关系图),并利用文献报道的方法拟合了他们的结合常数,以上结果均说明大环化合物11、化合物12均与c70分子有超分子相互作用。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。
