1.本发明涉及制药领域,具体地,涉及福多司坦的制备方法及产品。
背景技术:
2.福多司坦属于半胱氨酸衍生物,其结构式如式1所示,
[0003][0004]
福多司坦(fudosteine)化学名是(-)-(r)-2-氨基-3-(3-羟丙基硫代)丙酸,是由日本三菱制药株式会社与s.s制药株式会社共同研发的,2001年12月7日在日本上市。福多司坦是2017年国家医保药品目录新收载的药物,是一种新型的祛痰和黏痰溶解药物,该药适应症几乎涵盖包括支气管炎在内的所有呼吸系统疾病的多痰久咳性疾病。
[0005]
福多司坦对于慢性呼吸体系疾病起到了多种药理功能:阻止呼吸道上皮细胞增多,让痰液的海藻糖/唾液酸比例恢复常态,改善纤毛输送气道分泌液的水平,同时发挥抗炎功能。福多司坦属于一种新型的祛痰药物,相较于传统的氨溴索、乙酰半胱氨酸、羧甲司坦等,福多司坦因效果良好,低毒副作用,药物兼容性强等优势,逐渐成为了慢性呼吸体系疾病祛痰的主要药物之一,并用于治疗慢性喘息、慢性阻塞型肺气肿等呼吸体系疾病的祛痰阶段。
[0006]
目前,福多司坦的制备方法主要有5种:1、溴代醇取代法,利用l-半胱氨酸与3-溴-1-丙醇在氨(或其它有机碱)溶液中通过亲核取代反应制备,收率在80%左右;2、氧化还原自由基法,把l-半胱氨酸和烯丙醇通过过氧化物等催化剂引导下制备,收率91%;3、光催化法,在紫外照射下,l-半胱氨酸与丙烯醇可以高效地反应,收率与无紫外光照射相比能大幅提高,由40%提高到87.8%;4、微波法,将水、半胱氨酸、丙烯醇共同置于微波反应器内,设置反应温度及反应时长,待其反应完毕,用乙醇进行后处理,获得高纯度的福多司坦,收率92.74%;5、氧杂环丁烷法(参考专利:威海迪素制药有限公司.一种福多司坦的制备方法.cn 105461603 a.)。
[0007]
但是以上方法使用的原料价格昂贵,安全风险高,环保压力大,并且所制备的福多司坦收率不高,无机及有机杂质含量较高,因此研发一种新的成本低廉、环保、安全、收率高的福多司坦很有必要。
技术实现要素:
[0008]
本技术是基于发明人对以下事实和问题的发现和认识作出的:
[0009]
溴代醇取代法使用无机碱做缚酸剂会产生等量的无机盐,由于福多司坦水溶性较好,在醇析精制过程中无机盐不易除尽,导致产品铵盐、炙灼残渣和溴化物较高,同时l-半胱氨酸在碱性条件下容易发生氧化反应而产生胱氨酸杂质较高。另外,不论是氨还是有机碱,反应时间较长,成本较高,氨氮排放等环保压力不容忽视;溴代醇的价格昂贵,且来源有限。
[0010]
氧化还原自由基法单一应用无机类过氧化物引发剂,分解温度高,其剧烈的反应条件,会使得有关物质难以控制(特别是氧化反应产生的亚砜类杂质),影响其所得的药品品质。
[0011]
光催化法的生产设备价格昂贵,不利于推广。
[0012]
微波法在反应期间有蓝色未知物质产生,可能对产品品质造成影响,同时,大幅度过量使用剧毒的丙烯醇的风险也难以控制。
[0013]
氧杂环丁烷法使用氧杂环丁烷进行福多司坦的制备,氧杂环丁烷沸点、燃点很低,保存条件苛刻,安全风险很高。
[0014]
此外,上述方法使用半胱氨酸作为原料,成本较高,且稳定性差。
[0015]
此外,使用3-氯-1-丙醇做为反应原料,尽管成本较低,但是很难使得反应彻底完全,因而有较多的底物残留,以及底物转化而来的胱氨酸等杂质。
[0016]
本发明旨在至少在一定程度上解决相关技术中的技术问题之一。
[0017]
为此,在本发明的第一方面,本发明提出了一种福多司坦制备方法。根据本发明的实施例,所述方法包括:(1)将盐酸半胱氨酸与3-氯-1-丙醇在碱液中进行第一反应,得到福多司坦反应液;(2)将所述福多司坦反应液通过电渗析除杂进行第一纯化处理,得到所述福多司坦粗品;(3)将所述福多司坦粗品进行第二纯化处理,得到所述福多司坦精品。本发明提供的方法成本低,稳定性高,安全环保,福多司坦收率高,杂质低,使用本发明所制备的福多司坦晶体光泽度高、流动性好。
[0018]
根据本发明的实施例,上述方法还可进一步包括如下附加技术特征至少之一:
[0019]
根据本发明的实施例,所述电渗析除杂处理是在电渗析系统中进行的,当循环通电处理至淡室电导率降至不高于30ms/cm时,停止通电。根据本发明的实施例,电渗析可以高效的清除取代反应产生的大量无机盐,使得最终产品氯化物、重金属、残渣极低。
[0020]
根据本发明的实施例,所述电渗析除杂处理是在电渗析系统中进行的,当循环通电处理至淡室电导率降至10~30ms/cm时,停止通电。根据本发明的实施例,在淡室电导率降至10~30ms/cm时停止通电,可以达到较好的除杂效果,使无机盐、重金属、残渣等的含量降低。
[0021]
根据本发明的实施例,所述电渗析除杂处理是在电渗析系统中进行的,当循环通电处理至淡室电导率降至30ms/cm时,停止通电。根据本发明的实施例,在电导率为30ms/cm以下的时候停止通电是发明人经过反复试验所获得的最优反应条件,综合本发明的制备方法,淡室电导率至30ms/cm时大量杂质已被清除,仍有少量杂质残留于体系内,但若继续通电,福多司坦则会损失,考虑到后续仍有重结晶等除杂步骤,选择在此时停止通电,可以得到最佳的除杂效果,同时尽可能少损失产品
[0022]
根据本发明的实施例,所述碱液包括有机碱液和/或无机碱液,优选地,所述碱液包括氨水、氢氧化钠溶液和有机胺溶液的至少之一,更优选地,所述碱液为氢氧化钠溶液。根据本发明实施例的方法需要在碱性环境下进行,优选使用氢氧化钠成本更加低廉,反应速度更快,安全、环保压力更小,且无异味、无泄露风险。
[0023]
根据本发明的实施例,所述盐酸半胱氨酸与3-氯-1-丙醇的摩尔比为100:(100~120),优选为100:(105~115)。发明人经过大量的研究发现,采用上述配比可以提高生产效率,提高反应转化率,减少杂质生成。
[0024]
根据本发明的实施例,所述第一反应的ph值为7~9,温度为60~70℃。根据本发明的实施例,在上述ph条件下反应,可以提高反应效率,提高产率,此外,在上述温度条件下反应,反应的速率更快,利于成产,节约成本。
[0025]
根据本发明的实施例,进行步骤(2)之前,将所述福多司坦反应液与铜离子化合物进行混合处理,过滤,收集滤液,再将所述滤液进行步骤(3)的操作。根据本发明的实施例,加入铜离子可以在电渗析之前去除一部分杂质,使最终产物的纯度更高。
[0026]
根据本发明的实施例,所述铜离子化合物选自硫酸铜。
[0027]
根据本发明的实施例,进行所述混合处理之前,预先调节所述福多司坦反应液的ph值至5~6。根据本发明的实施例,在进行所述混合处理之前,将福多司坦反应液的ph调节至福多司坦的等电点,可以避免电渗析过程中福多司坦的浪费,有利于去除杂质。
[0028]
根据本发明的实施例,铜离子化合物用量为所述盐酸半胱氨酸摩尔量的1/1000~1/2000。
[0029]
根据本发明的实施例,所述铜离子化合物是以水溶液的形式提供的,浓度为0.01mol/l。
[0030]
根据本发明的实施例,上述铜离子的添加量既可以不损伤膜系统,又可以在不残留过多的前提下消除半胱氨酸、胱氨酸等杂质。
[0031]
根据本发明的实施例,所述混合处理的时间为1~1.5小时,混合结束后溶液的温度为20~35℃。发明人发现,采用边反应边降温的方式,在絮凝杂质的过程中对液体进行降温,有利于后续的电渗析处理,同时可以节约反应时间,降低成本。
[0032]
根据本发明的实施例,进行所述电渗析除杂处理之后,将得到电渗析除杂处理产物进行重结晶,过滤,收集结晶产物,得到所述福多司坦。根据本发明的实施例,在电渗析处理后,进一步对处理后的福多司坦粗品进行重结晶处理,除去杂质,所得福多司坦的杂质含量更少,纯度更高。
[0033]
根据本发明的实施例,进行所述重结晶之后,将所得结晶产物与乙醇溶液进行打浆处理,得到所述福多司坦,其中,所述打浆处理的时间为1~1.5小时。发明人经过反复试验发现,利用95%的乙醇对所述结晶产物进行打浆,可以去除有机含氯化合物,降低产物的基因毒性杂质,所得福多司坦晶体的光泽度高、分散性好、流动性好。
[0034]
根据本发明的实施例,所述盐酸半胱氨酸以盐酸半胱氨酸无水物和/或盐酸半胱氨酸水合物的形式提供;优选地,所述盐酸半胱氨酸以盐酸半胱氨酸一水物的形式提供。根据本发明实施例的方法,使用盐酸半胱氨酸无水物形式或盐酸半胱氨酸水合物的形式制备福多司坦均可获得较好的收率,发明人发现使用盐酸半胱氨酸一水物更加稳定,且成本更低,同时也可以具有良好的福多司坦收率,杂质含量也符合国家标准,因此优选盐酸半胱氨
酸一水物的形式制备福多司坦。
[0035]
在本发明的第二方面,本发明提出了一种福多司坦制品,所述福多司坦制品是根据前面所述福多司坦制备方法制备的。根据本发明的实施例,所述福多司坦中胱氨酸等杂质含量极低,氯化物、硫酸盐、铵盐、残渣等无机杂质含量极低,福多司坦晶体的光泽度高、分散性和流动性好。
[0036]
本发明的附加方面和优点将在下面的描述中部分给出,部分将从下面的描述中变得明显,或通过本发明的实践了解到。
附图说明
[0037]
本发明的上述和/或附加的方面和优点从结合下面附图对实施例的描述中将变得明显和容易理解,其中:
[0038]
图1是根据本发明实施例1的福多司坦产品的hplc图谱;
[0039]
图2是根据本发明对比例1的福多司坦产品的hplc图谱;
[0040]
图3根据本发明实施例1的福多司坦产品的红外图谱。
具体实施方式
[0041]
下面详细描述本发明的实施例,所述实施例的示例在附图中示出,下面通过参考附图描述的实施例是示例性的,旨在用于解释本发明,而不能理解为对本发明的限制。
[0042]
此外,术语“第一”、“第二”仅用于描述目的,而不能理解为指示或暗示相对重要性或者隐含指明所指示的技术特征的数量。由此,限定有“第一”、“第二”的特征可以明示或者隐含地包括至少一个该特征。在本发明的描述中,“多个”的含义是至少两个,例如两个,三个等,除非另有明确具体的限定。
[0043]
福多司坦制备方法
[0044]
在本发明的一个方面,本发明提出了一种福多司坦制备方法。根据本发明的实施例,所述方法包括:(1)将盐酸半胱氨酸与3-氯-1-丙醇在碱液中进行第一反应,得到福多司坦反应液;(2)将所述福多司坦反应液通过电渗析除杂进行第一纯化处理,得到所述福多司坦粗品;(3)将所述福多司坦粗品进行第二纯化处理,得到所述福多司坦精品。所述纯化处理包括:将所述福多司坦进行电渗析脱盐处理。根据本发明实施例的方法使用价格更加低廉、稳定性更高的盐酸半胱氨酸替代半胱氨酸,降低了成本,盐酸半胱氨酸可以以盐酸半胱氨酸无水物和/或盐酸半胱氨酸水合物的形式提供,优选为盐酸半胱氨酸一水物,盐酸半胱氨酸一水物更加稳定,对制备环境的要求更低,进一步降低了生产成本,有利于大规模生产。并且,反应过程使用的3-氯-1-丙醇,比剧毒的烯丙醇、易燃易爆的氧杂环丁烷更加安全,价格更低廉。
[0045]
在一个实施例中,所述电渗析脱盐处理是在电渗析系统中进行的,当循环通电处理至淡室电导率降至30ms/cm时,停止通电。将待处理的溶液导入电渗析系统淡室进行脱盐,电渗析系统浓室加入等体积的水,以均相膜为媒介,极室以稀硫酸和稀氢氧化钠溶液为极液,循环通电,至淡室电导率降至30ms/cm以下,停止通电。淡室放料,料液在60~70℃减压浓缩至原体积的20%,流加2~3倍体积的95%乙醇,降温至-4℃,析晶4~5小时,离心甩料得到待精制的福多司坦产品。在电导率为30ms/cm以下的时候停止通电是发明人经过反
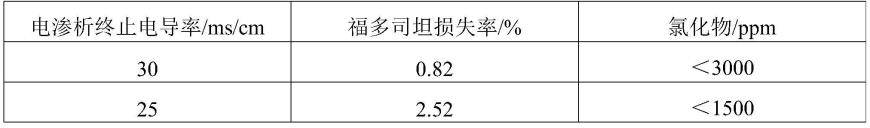
复试验所获得的最优反应条件,综合本发明的制备方法,淡室电导率至30ms/cm时大量杂质已被清除,仍有少量杂质残留于体系内,但若继续通电,福多司坦则会损失,考虑到后续仍有重结晶等除杂步骤,选择在此时停止通电,可以得到最佳的除杂效果,同时尽可能少损失产品,发明人经过反复实验,得到电渗析终点电导率、产品损失率和氯离子对应关系,对应关系见表1。
[0046]
表1:电渗析终点电导率、产品损失率和氯离子对应表
[0047][0048][0049]
在一个优选的实施例中,上述流加乙醇结晶过程中,既可以使用乙醇,也可以使用甲醇。流加乙醇或甲醇的温度为50℃,在该温度下可以保证晶体析出结晶,同时减少杂质的析出,使所得晶体的纯度更高。
[0050]
在一个具体的实施例中,在充氮保护的反应容器内,将1倍质量的盐酸半胱氨酸一水物溶于2倍体积纯化水后,加入0.56~0.65倍质量的3-氯-1-丙醇,缓慢流加或滴加入1.9倍质量的30%的氢氧化钠溶液,加料完毕后,升温,控制温度在60~70℃左右。以碘量法监控反应进程,反应完成后,用盐酸调节ph至5~6。
[0051]
在一个具体的实施例中,向反应液中加入盐酸半胱氨酸一水物的0.05%~0.2%倍质量的硫酸铜(配制成0.2%的水溶液)。1~1.5小时内逐渐降温至20~35℃,搅拌,絮凝,保持充氮状态。混合处理1~1.5小时可使前述反应过程中生成的胱氨酸、半胱氨酸等杂质充分絮凝,以便从溶液中析出去除。溶液需要降温至20~35℃才可进行电渗析处理,否则会损坏电渗析处理装置。
[0052]
在本发明的一个实施例,3-氯-1-丙醇活性不是很强,与半胱氨酸反应很难彻底完全,因而会残留一定含量的半胱氨酸,而半胱氨酸在合成过程的碱性条件下,强烈地氧化成为胱氨酸。铜离子与巯基/二硫键有特殊的化学亲和力,加入微量硫酸铜,可以促进胱氨酸(胱氨酸溶解度很小,是由盐酸半胱氨酸一水物或半胱氨酸氧化而成)絮凝析出,从而在电渗析前的过滤步骤,被大部分清除;另外,盐酸半胱氨酸(起始物料)、二羟丙基二硫醚(碱降解杂质)等杂质,使其形成铜络离子形态(半胱氨酸剩余较多的时候会与铜离子形成部分沉淀,在电渗析过程前被过滤掉),在电渗析过程会受电场影响,从淡室迁移到浓室,从而可以与福多司坦分离;多余的硫酸铜和硫酸盐会在电渗析过程中,受电场影响而清除,不影响产品质量。所用的硫酸铜可以是无水物,也可以是含水物。
[0053]
在本发明的一个实施例中,所述重结晶的方法是:脱色容器中加入福多司坦粗品,并加入粗品重量0.8~1.5倍的纯化水。搅拌加热至60~70℃溶解,加入福多司坦固体量0.5%的活性炭脱色20分钟,保温过滤,滤液降至50℃后,流加溶液体积4~6倍95%乙醇,加完后降温至-4℃,析晶4~5小时。离心甩料,所得半成品,用2~5倍的95%乙醇室温打浆1~
1.5小时。离心甩料,60℃下真空干燥,粉碎粉筛得到成品福多司坦。上述室温打浆过程,不仅可以进一步去除具有基因毒性杂质,也可以使所得产品的光泽度、分散性、流动性得到较大提升。
[0054]
本发明所提出的制备福多司坦的方法具有下述优点:
[0055]
1、所得产品的胱氨酸等杂质含量极低。
[0056]
2、所得产品的氯化物、硫酸盐、铵盐、残渣等无机杂质极低。
[0057]
3、所得产品的光泽、分散性和流动性好。
[0058]
4、不使用剧毒的烯丙醇或者高危的氧杂环丁烷。
[0059]
5、收率较高,连续生产时,收率更高。
[0060]
下面参考具体实施例,对本发明进行描述,需要说明的是,这些实施例仅仅是描述性的,而不以任何方式限制本发明。
[0061]
实施例1
[0062]
步骤1、在充氮容器内,将100克盐酸半胱氨酸一水物溶于200毫升水中,溶清后加入59.3克3-氯-1-丙醇,搅拌均匀。
[0063]
步骤2、0.5小时内滴加190克30%的氢氧化钠溶液。
[0064]
步骤3、升温至65℃,并保温。
[0065]
步骤4、1小时后,以碘量法监控终点,3滴碘液(0.1mol/l)深蓝。
[0066]
步骤5、加入工业盐酸调节溶液ph=5.72。
[0067]
步骤6、加入25克硫酸铜0.2%的水溶液,继续搅拌,降温至35℃。
[0068]
步骤7、过滤,滤液导入电渗析系统。
[0069]
步骤8、通电至电导率降至30ms/cm,停止通电。
[0070]
步骤9、淡室放料,约400毫升,65℃减压浓缩至约80毫升。
[0071]
步骤10、冷至50℃,流加95%乙醇250毫升。搅拌降温至-4℃,析晶4小时。
[0072]
步骤11、离心甩料,得到粗品116克,含水量15%。
[0073]
步骤12、粗品加入120克纯化水,65℃加热溶解,加0.5克活性炭,保温脱色20分钟。过滤。
[0074]
步骤13、滤液约200毫升,降温至50℃,流加乙醇1200毫升,加完后降温至-4℃。析晶4小时。离心甩料得到半成品107克,含水量12%。
[0075]
步骤14、半成品加入300毫升95%乙醇,打浆1小时。
[0076]
步骤15、离心甩料,60℃真空干燥至恒重,粉碎粉筛,得到成品福多司坦93克。收率93%。
[0077]
所获福多司坦液相色谱检测结果及红外光谱结果如图1和图3所示。
[0078]
实施例2
[0079]
步骤1、在充氮容器内,将100克盐酸半胱氨酸一水物溶于200毫升水中,溶清后加入59.3克3-氯-1-丙醇,搅拌均匀。
[0080]
步骤2、0.5小时内滴加190克30%的氢氧化钠溶液。
[0081]
步骤3、升温至65℃,并保温。
[0082]
步骤4、1小时后,以碘量法监控终点,3滴碘液(0.1mol/l)深蓝。
[0083]
步骤5、加入工业盐酸调节溶液ph=5.60。
[0084]
步骤6、加入25克硫酸铜0.2%的水溶液,继续搅拌,降温至35℃。
[0085]
步骤7、过滤,滤液导入电渗析系统。
[0086]
步骤8、通电至电导率降至30ms/cm,停止通电。
[0087]
步骤9、淡室放料,约400毫升。淡室放空后用纯化水,分两次,每次200毫升淡室内循环5分钟,放出。淡室放料和淡室洗水合并约800毫升,65℃减压浓缩至约80毫升。
[0088]
步骤10、冷至50℃,流加95%乙醇250毫升。搅拌降温至-4℃,析晶4小时。
[0089]
步骤11、离心甩料,得到粗品120克,含水量16%。
[0090]
步骤12、粗品加入120克纯化水,65℃加热溶解,加0.5克活性炭,保温脱色20分钟。过滤。
[0091]
步骤13、滤液约200毫升,降温至50℃,流加乙醇1200毫升,加完后降温至-4℃。析晶4小时。离心甩料得到半成品109克,含水量11%。
[0092]
步骤14、半成品加入300毫升95%乙醇,打浆1小时。
[0093]
步骤15、离心甩料,60℃真空干燥至恒重,粉碎粉筛,得到成品福多司坦96克。收率96%。
[0094]
实施例3
[0095]
步骤1、1000l反应釜内,将100公斤盐酸半胱氨酸一水物溶于200升水中,溶清后加入59.3公斤3-氯-1-丙醇,搅拌均匀,抽成真空。
[0096]
步骤2、1小时内滴加190公斤30%的氢氧化钠溶液。
[0097]
步骤3、打开蒸汽阀,升温至65℃,并保温。
[0098]
步骤4、1小时后,以碘量法监控终点,2滴碘液(0.1mol/l)深蓝。
[0099]
步骤5、加入工业盐酸调节溶液ph=5.82。
[0100]
步骤6、加入25公斤硫酸铜0.2%的水溶液,继续搅拌,降温至35℃。
[0101]
步骤7、过滤,滤液导入电渗析系统。
[0102]
步骤8、通电至电导率降至30ms/cm,停止通电。
[0103]
步骤9、淡室放料,约400升,65℃减压浓缩至约80升。
[0104]
步骤10、冷至50℃,流加95%乙醇250升。搅拌降温至-4℃,析晶4小时。
[0105]
步骤11、离心甩料,得到粗品114公斤,含水量16%。
[0106]
步骤12、粗品投入脱色罐中,加入120升纯化水,65℃加热溶解,加0.5公斤活性炭,保温脱色20分钟。板框过滤,再过精滤器。
[0107]
步骤13、滤液约200升,降温至50℃,流加乙醇1200升,加完后降温至-4℃。析晶4小时。离心甩料得到半成品106公斤,含水量13%。
[0108]
步骤14、半成品加入300升95%乙醇,打浆1小时。
[0109]
步骤15、离心甩料,60℃真空干燥至恒重,粉碎粉筛,得到成品福多司坦92.9公斤。收率92.9%。
[0110]
对比例1未加入铜离子
[0111]
步骤1、在充氮容器内,将100克盐酸半胱氨酸一水物溶于200毫升水中,溶清后加入59.3克3-氯-1-丙醇,搅拌均匀。
[0112]
步骤2、0.5小时内滴加190克30%的氢氧化钠溶液。
[0113]
步骤3、升温至65℃,并保温。
[0114]
步骤4、1小时后,以碘量法监控终点,3滴碘液(0.1mol/l)深蓝。
[0115]
步骤5、加入工业盐酸调节溶液ph=5.54。
[0116]
步骤6、过滤,滤液导入电渗析系统。
[0117]
步骤7、通电至电导率降至30ms/cm,停止通电。
[0118]
步骤8、淡室放料,约400毫升,65℃减压浓缩至约80毫升。
[0119]
步骤9、冷至50℃,流加95%乙醇250毫升。搅拌降温至-4℃,析晶4小时。
[0120]
步骤10、离心甩料,得到粗品116克,含水量15%。
[0121]
步骤11、粗品加入120克纯化水,65℃加热溶解,加0.5克活性炭,保温脱色20分钟。过滤。
[0122]
步骤12、滤液约200毫升,降温至50℃,流加乙醇1200毫升,加完后降温至-4℃。析晶4小时。离心甩料得到半成品107克,含水量12%。
[0123]
步骤13、半成品加入300毫升95%乙醇,打浆1小时。
[0124]
步骤14、离心甩料,60℃真空干燥至恒重,粉碎粉筛,得到成品福多司坦92.1克。收率92.1%。
[0125]
所述福多司坦的的液相色谱检测结果如图2所示。
[0126]
对比例2未使用铜离子,未进行电渗析处理,碱液为氨水
[0127]
步骤1、在充氮容器内,将100克盐酸半胱氨酸一水物溶于200毫升水中,溶清后加入59.3克3-氯-1-丙醇,搅拌均匀。
[0128]
步骤2、用25%氨水调节ph 8.50。
[0129]
步骤3、升温至65℃,并保温。
[0130]
步骤4、3小时后,以碘量法监控终点,3滴碘液(0.1mol/l)深蓝。
[0131]
步骤5、加入工业盐酸调节溶液ph=5.72。
[0132]
步骤9、反应液约400毫升,65℃减压浓缩至约80毫升。
[0133]
步骤10、冷至50℃,流加95%乙醇250毫升。搅拌降温至-4℃,析晶4小时。
[0134]
步骤11、离心甩料,得到粗品106克,含水量21%。
[0135]
步骤12、粗品加入100克纯化水,65℃加热溶解,加0.5克活性炭,保温脱色20分钟。过滤。
[0136]
步骤13、滤液约200毫升,降温至50℃,流加乙醇1200毫升,加完后降温至-4℃。析晶4小时。离心甩料得到半成品101克,含水量17%。
[0137]
步骤14、半成品加入300毫升95%乙醇,打浆1小时。
[0138]
步骤15、离心甩料,60℃真空干燥至恒重,粉碎粉筛,得到成品福多司坦82.8克。收率82.8%。
[0139]
对比例3未加入铜离子,未进行电渗析
[0140]
步骤1、在充氮容器内,将100克盐酸半胱氨酸一水物溶于200毫升水中,溶清后加入59.3克3-氯-1-丙醇,搅拌均匀。
[0141]
步骤2、0.5小时内滴加190克30%的氢氧化钠溶液。
[0142]
步骤3、升温至65℃,并保温。
[0143]
步骤4、1小时后,以碘量法监控终点,3滴碘液(0.1mol/l)深蓝。
[0144]
步骤5、加入工业盐酸调节溶液ph=5.63。
[0145]
步骤6、反应液约400毫升,65℃减压浓缩至约80毫升,过滤脱盐。
[0146]
步骤9、滤液冷至50℃,流加95%乙醇250毫升。搅拌降温至-4℃,析晶4小时。
[0147]
步骤10、离心甩料,得到粗品102克,含水量21%。
[0148]
步骤11、粗品加入100克纯化水,65℃加热溶解,加0.5克活性炭,保温脱色20分钟。过滤。
[0149]
步骤12、滤液约200毫升,降温至50℃,流加乙醇1200毫升,加完后降温至-4℃。析晶4小时。离心甩料得到半成品95克,含水量19%。
[0150]
步骤13、半成品加入300毫升95%乙醇,打浆1小时。
[0151]
步骤14、离心甩料,60℃真空干燥至恒重,粉碎粉筛,得到成品福多司坦73.5克。收率75.5%。
[0152]
对比例4未加入乙醇打浆
[0153]
步骤1~13与实施例1相同,步骤13所得半成品,不打浆,直接干燥并粉碎,以获得福多司坦成品。
[0154]
实施例与对比例检测结果
[0155]
表2:实施例和对比例相关物质(hplc)检测结果比较
[0156][0157]
检测方法为jp17标准。从以上结果可以看出,实施例中杂质的含量低于各对比例。
[0158]
表3:实施例和对比例的理化检测结果比较
[0159][0160]
上述各指标检测方法为jp17标准。从以上结果可以看出,采用本发明制备的福多司坦,无机杂质含量显著低于各对比例。
[0161]
表4:实施例和对比例的收率比较
[0162][0163][0164]
从以上结果可以看出,采用本发明制备的福多司坦,收率优于各对比例。
[0165]
由于电渗析系统内会残留部分产品,料液排空后用水循环洗出残留产品可以相应提高收率(实施例2)。连续生产时,可以不洗涤电渗析系统内部,残留产品会直接加入到下一个批次,相应提高收率。
[0166]
表5:实施例的其它检测数据
[0167][0168]
表6:休止角测试
[0169]
测试样品休止角/
°
实施例152对比例429
[0170]
由上表可以看出,对比例4未经打浆,产品直接粉碎烘干后通过固定漏斗法测得其休止角为52
°
,流动性较差;而实施例1后通过固定漏斗法测得其休止角为29
°
,流动性优良。
[0171]
采用本发明制备的福多司坦,产品品质可以满足jp17等药品标准,并且由于采用了价格更加低廉的盐酸半胱氨酸一水物和3-氯-1-丙醇,配合络合-电渗析除杂工艺,物料/能耗等方面成本大幅度降低,同时也避免了氨氮排放的环保压力。
[0172]
在本说明书的描述中,参考术语“一个实施例”、“一些实施例”、“示例”、“具体示例”、或“一些示例”等的描述意指结合该实施例或示例描述的具体特征、结构、材料或者特点包含于本发明的至少一个实施例或示例中。在本说明书中,对上述术语的示意性表述不必须针对的是相同的实施例或示例。而且,描述的具体特征、结构、材料或者特点可以在任一个或多个实施例或示例中以合适的方式结合。此外,在不相互矛盾的情况下,本领域的技术人员可以将本说明书中描述的不同实施例或示例以及不同实施例或示例的特征进行结合和组合。
[0173]
尽管上面已经示出和描述了本发明的实施例,可以理解的是,上述实施例是示例性的,不能理解为对本发明的限制,本领域的普通技术人员在本发明的范围内可以对上述实施例进行变化、修改、替换和变型。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。