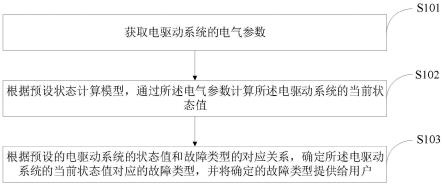
1.本发明涉及分析化学和代谢组学领域,是一种基于分子结构关联网络的规模化代谢组定性方法。
2.研究背景
3.代谢组学(nicholson,jk et al.,metabonomics:a platform for studying drug toxicity and gene function.nature reviews drug discovery 2002,1,153-161.)是以生物体中分子量小于1000的小分子代谢物为研究对象,运用多种分析手段如高分辨质谱(hrms)、核磁共振(nmr)、色谱-质谱联用等从整体水平上研究体内代谢物组成及其与生理病理相关的变化规律。代谢组学已被广泛应用于生命科学、疾病诊断、药物安全性评价、药理研究、营养科学等多个领域。
4.代谢物结构鉴定是代谢组学发展的瓶颈问题之一。生物体内代谢物成分及其复杂、理化性质各异、含量差别巨大,生物样品中的代谢物浓度分布范围达7~9个数量级。随着分析技术的不断进步,特别是高分辨质谱技术迅猛发展,生物样本非靶向代谢组学分析获得了海量的代谢组数据,但其中可被鉴定的代谢物不超过25%(baker,m.,metabolomics:from small molecules to big ideas.nature methods 2011,8,117-121.)。质谱数据库检索是代谢物鉴定的最主要方式,其收录情况直接决定了代谢物鉴定的数量与质量。metlin数据库目前已收录超过1000,000种化合物,其中50万多种有二级质谱(ms/ms)数据;人类代谢组数据库(hmdb)(http://www.hmdb.ca/)收录114,260种化合物,仅有7,418多个化合物有实测ms/ms谱图数据,279,972种为预测ms/ms数据。预测二级谱图是解决二级短缺问题的一种手段,但大多预测谱图与实验谱图相差很大,仍然会给代谢物的定性带来困难。基于实验二级质谱相似性的分子网络如gnps(https://gnps.ucsd.edu/)已被证明是一种有效的代谢物辅助定性工具。其基于具有相似二级谱图的代谢物其结构具有相似性的假设,但采用该方法对代谢物结构鉴定仍需依赖大规模的实验二级谱图数据库,因此其定性能力仍受到目前代谢组数据库实验二级谱图短缺的制约。
5.目前代谢组学研究主要基于其中的已知代谢物,大量稳定、可重复检测的“未知代谢物”虽可能具有重要生物功能,却因无法鉴定分子结构,未能对其利用或开展深入研究,导致质谱信息利用率低。因此,亟待发展功能代谢组学新技术、新方法,包括更高效可靠的算法、知识库及相关工具来实现对非靶向超高效液相色谱-高分辨质谱(uhplc-hrms)代谢谱数据的深度解析。因此,发展能够辅助代谢物结构解析,尤其是能够鉴定未知代谢物的分析新方法显得尤为迫切。
6.为此,本发明充分利用代谢组数据库中丰富的一级及其结构信息,发展一种不依赖大规模二级数据库的基于分子结构关联网络的规模化代谢组定性方法,突破保留时间和二级信息缺乏造成的定性瓶颈。
技术实现要素:
7.本发明提供了一种基于分子结构关联网络的代谢组规模化定性方法。为了实现本
发明目的,收集开源代谢组数据库中的内源性代谢物,基于所获取的代谢物分子指纹相似性构建代谢物分子结构关联网络;对生物样本提取物进行基于超高效液相色谱-高分辨质谱的非靶向代谢组学分析,获取生物样本提取物的代谢组相关色谱-质谱信息;基于标样的分子描述符和实验保留时间,构建保留时间预测模型;利用保留时间预测模型计算分子结构关联网络中每个节点的预测保留时间;利用非靶向超高效液相色谱-高分辨质谱代谢组实验数据,以分子结构关联网络作为背景网络,进行代谢组规模化定性。本发明采用的技术方案步骤如下:
8.第一步,构建代谢组的分子结构关联网络;根据开源代谢组数据库如人类代谢组hmdb数据库、京都基因与基因组百科全书kegg数据库获取内源性代谢物,包括其名称,单同位素质量,分子结构数据文件;基于其分子结构数据文件获得每个代谢物的分子指纹,这里分子指纹可以是圆形指纹、maccs指纹、原子对指纹和拓扑指纹中的任意一种;计算任意两个候选代谢物分子指纹间的相似度;设定相似性阈值,以代谢物为节点,分子指纹相似性为边,构建代谢组水平的分子结构关联网络;
9.第二步,采用超高效液相色谱-高分辨质谱对待测生物样本的提取物进行非靶向代谢组学分析;获取提取物代谢组的相关色谱-质谱信息,包括实验测得的代谢物峰的保留时间t
r实测
,一级质谱信息即一级离子质荷比m/z
实测
,和相应的二级质谱信息即二级离子的质荷比和强度;一级离子指化合物被离子化后直接采集到的离子;二级离子指一级离子在施加一定能量碰撞碎裂后采集到的离子;
10.第三步,构建保留时间预测模型;采用与生物提取物相同的超高效液相色谱-高分辨质谱数据采集条件,分析内源性代谢物的混合标样,获取其液相色谱实验保留时间;采用开源网站chemdes(http://www.scbdd.com/chemdes)和dragon7(https://chm.kode-solutions.net/index.php)获取标样的分子描述符;基于实验保留时间和分子描述符,通过定量结构-保留关系构建保留时间预测模型;保留时间预测模型采用多重线性回归模型、弹性网络回归模型、随机森林模型、支持向量机模型和深度学习模型中任意一种方法建立;
11.第四步,计算分子结构关联网络中代谢物的预测保留时间;基于其分子结构数据文件获得每个代谢物的分子描述符;利用保留时间预测模型计算分子结构关联网络中每个节点的预测保留时间;
12.第五步,基于分子结构关联网络对非靶向代谢组学数据进行规模化定性;以第一步构建的分子结构关联网络作为背景网络,利用标准样品从非靶向超高效液相色谱-高分辨质谱代谢组实验数据鉴定50~100个代谢物作为种子代谢物,将其映射到已建立的分子结构关联网络中,从网络中获取种子代谢物的相邻代谢物,这里相邻代谢物指分子结构关联网络中有直接边连接的代谢物;将种子代谢物的二级质谱作为相邻代谢代谢物二级质谱,设定搜索阈值,在实验数据中搜索与相邻代谢物m/z
理论
,t
r预测
,二级质谱匹配的代谢物峰,匹配成功则该代谢物峰鉴定完成;当有多个匹配结果时,对匹配结果进行打分;鉴定出的代谢物再作为新的种子,重复定性过程,直至没有新的代谢物被定性出来。
13.本发明依据结构相似的代谢物,其ms/ms具有相似性这一前提,建立了代谢组数据库导向的基于分子结构关联网络的规模化定性方法,实现对未知代谢物的结构鉴定。通过建立代谢物数据库及其代谢物分子结构关联网络,采用分子结构关联网络去鉴定没有标准ms/ms谱图的代谢物,使得代谢物的结构鉴定可不依赖大规模的标准ms/ms数据库。本发明
是一种不依赖于大规模实验二级谱图数据库的代谢组深度注释方法,可实现代谢组规模化、可靠定性,显著扩大代谢组注释的覆盖度。
附图说明
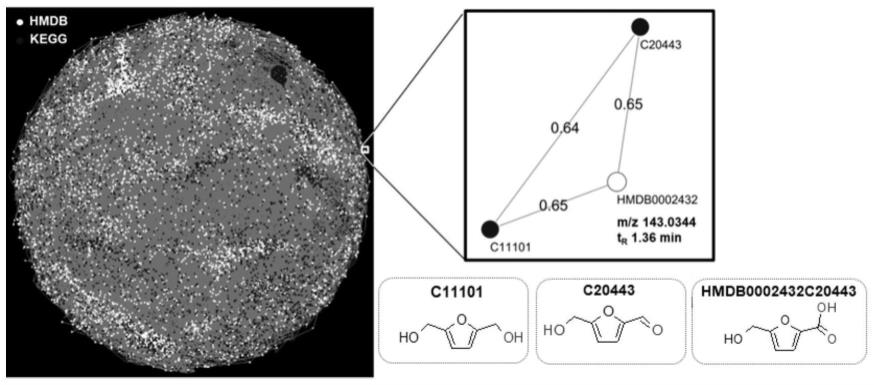
14.图1正离子模式分子结构关联网络及其局部放大示意图;
15.图2基于分子结构关联网络的代谢物定性过程示意图;
16.图3正负离子模式下小鼠肝脏的鉴定结果(共鉴定518个代谢物);
17.图4分子结构关联网络定性方法与现有方法对小鼠肝脏组织代谢物的鉴定结果比较。
具体实施方式
18.下面结合附图对本发明实施作详细说明:本实施例在以本发明技术方案为前提下进行实施,给出了详细的实施方式和具体的操作过程,但本发明的保护范围不限于下述的实施例。
19.实施例1
20.为了证实本发明的有效性和可行性,在组织提取物中添加由885个代谢物(包括尿嘧啶核苷和嘌呤等代谢物,添加终浓度为0.1~5微克/毫升)组成的混合标样,混合标样为从构建的分子结构网络中选取的,有市售标准品的代谢物组成。
21.组织代谢组的提取:采用动物代谢组学方法,提取小鼠肝脏中的代谢物。首先称取10毫克小鼠肝脏组织于1.5毫升离心管中,加入600微升的甲醇/水(体积比,4:1)提取剂(含由885个代谢物组成的混合标样),研磨珠打进行匀浆处理(25hz
×
1min*2),在4℃,12000rpm条件下离心15min。取400μl上清液,放入真空离心浓缩仪冻干。在冻干样品粉末中加入50微升乙腈/水(体积比,1:3),涡旋1min,在高速离心机中采用4摄氏度,14000rpm转速,离心10分钟。
22.将加标后的生物样本提取物进行超高效液相色谱-高分辨质谱数据采集,以采集的非靶向代谢组学数据中混合标样的定性为例进行发明的原理性说明。
23.基于代谢组数据库的分子结构关联网络构建:分子结构关联网络中的代谢物来自开源代谢组数据库人类代谢组数据库(hmdb)(https://hmdb.ca/)和京都基因与基因组百科全书数据库(kegg)(https://www.genome.jp/kegg/)。以正离子模式为例,初始代谢物源自hmdb数据库收录的5170个代谢物以及kegg数据库收录的5102个代谢物,并从上述数据库中获取每个代谢物的名称,单同位素质量及分子结构数据文件。由分子结构数据文件计算每个代谢物的分子指纹,本例中采用圆形指纹,计算任意两个分子圆形指纹间的相似度(相似度计算方法来源于开源工具rdkit)。设置分子指纹相似度阈值为0.6,符合该阈值的代谢物共有8562个(4141个代谢物来自hmdb,4421个代谢物来自kegg)。以代谢物为节点,分子间指纹相似性为边,构建分子结构关联网络(共有8,562个节点和264,095条边),正离子模式下的分子结构关联网络见图1(左图)。以代谢物hmdb0002432(hmdb数据库id)为例,其名称为5-羟甲基糠酸(5-hydroxymethyl-2-furoate),其分子结构见图1(右下图);它的电喷雾质谱正离子模式下[m h]
的m/z
理论
,t
r预测
分别为m/z 143.0344,1.36min。将代谢物hmdb0002432及其周边局部放大见图1(右上图),分子结构关联网络中有直接边连接的代谢
物称之为相邻代谢物,hmdb0002432的两个相邻代谢物分别为5-羟甲基呋喃醛(5-hydroxymethylfurfural,kegg数据库id:c11101)和5-羟甲基呋喃醇(5-hydroxymethylfurfuralalcohol,kegg数据库id:c20443)。从图中三个代谢物的分子结构可知,它们均具有5-羟甲基呋喃子结构,仅在2位的取代基有差异,分别为醛基,醇基和羧基。因此,其分子结构均有较好的相似性,其中hmdb0002432与c11101和c20443的指纹相似度均为0.65,c11101和c20443的指纹相似度为0.64,均满足相似度阈值,故在网络中有直接连边。
[0024]
实验非靶向色谱-质谱信息获取:数据采集于acquity uhplc超高效液相色谱系统(uplc,waters,milford,ma,usa.)和q exactive hf高分辨质谱(thermo fisher scientific,rockford,il,usa.)联用的分析仪器。
[0025]
正离子模式下液相色谱条件为:a相和b相分别为0.1%的甲酸/水(体积比)和0.1%的甲酸/乙腈(体积比)。流速为0.35ml/min。总分析时间为30min。洗脱梯度以5%b开始,保持1min。然后在23min内线性增加到100%b,并保持4min。接着在0.1min内线性回到初始比例,并保持2.9min。样品采用acquity beh c8色谱柱(100mm x 2.1mm,1.7μm,waters,milford,ma,u.s.a.)进行分离。柱温为50℃。样品进样室温度设置为4℃,样品进样量为5μl。
[0026]
负离子模式下液相色谱条件为:a相和b相分别为6.5mm碳酸氢铵-水和6.5mm碳酸氢铵-95%甲醇/水(体积比)。流速为0.35ml/min。总分析时间为25min。洗脱梯度以2%b开始,1min后开始线性增长,在18min时到100%b,并保持到22min。接着以线性方式在22.1min时回到初始比例,并保持到25min。样品采用acquity hss t3色谱柱(100mm x 2.1mm,1.8μm,waters,milford,ma,u.s.a.)进行分离。柱温为50℃。样品进样室温度设置为4℃,样品进样量为5μl。
[0027]
q exactive hf质谱分析条件为:扫描模式为全扫加二级质谱采集模式(full ms/data-dependent(ddms2))。在全扫质谱设置中,分辨率采用120,000。自动增益控制目标(agc target)和最大注入时间(maximum it)分别设置为3
×
106离子容量和100ms。全扫质量扫描范围为m/z 85-1250。在二级质谱设置中,自动增益控制目标(agc target)和最大注入时间(maximum it)分别设置为1
×
105离子容量和50ms。隔离窗口为m/z 1.0。碰撞能采用混合归一化能量15%,30%和45%。二级的采集由每个全扫描循环中响应最强的前10个离子触发。加入inclusion list并设置为on。正、负离子模式下喷雾电压分别为3.5kv和3.0kv,离子传输管的温度为320℃,辅助气的加热器温度为350℃。鞘气和辅助气流速分别为45和10(in arbitrary units)。s-lens设置为50.0(in arbitrary units)。
[0028]
在采集到的非靶向代谢组学数据中,提取出885个代谢物的实验保留时间t
r实测
,一级质谱信息即一级离子质荷比m/z
实测
和相应的二级质谱信息即二级离子的质荷比和强度。
[0029]
保留时间预测模型构建:采用与动物提取物相同的超高效液相色谱-高分辨质谱数据采集条件,分析1551个标样,获取液相色谱实验保留时间。利用标样的sdf文件在开源网站chemdes(http://www.scbdd.com/chemdes)中计算获得每个标样的1d&2d分子描述符,采用随机森林方法,构建保留时间预测模型。
[0030]
基于分子结构关联网络定性:以构建的分子结构关联网络作为背景网络,对非靶向超高效液相色谱-高分辨质谱代谢组采集的加标代谢物进行鉴定。以正离子模式为例,定
性的具体过程如下:
[0031]
1)利用标准样品从加标的组织提取物非靶向超高效液相色谱-高分辨质谱代谢组实验数据中鉴定出60个代谢物作为种子代谢物,将其映射到已建立的分子结构关联网络中,从网络中获取种子代谢物相邻代谢物。如图2所示,种子代谢物hmdb0036565(β-紫罗兰酮,beta-ionone)的相邻代谢物共有4个,分别为hmdb0032913(二羟基-β-紫罗兰酮,dihydro-β-ionone)、c02110(视黄酸,9-cis-retinoic acid)、hmdb0059883(α-紫罗兰酮,alpha-ionone)和hmdb0041011(β-环高柠檬醛,beta-cyclocitral)。
[0032]
2)将种子代谢物的二级质谱赋给相邻代谢代谢物,作为其拟二级质谱。设定搜索阈值,实验数据中代谢物峰的一级离子质荷比m/z
实测
,实验保留时间t
r实测
与相邻代谢物m/z
理论
,t
r预测
匹配的阈值同时满足|t
r预测-t
r实测
|/t
r预测
*100%《30%与|m/z
理论-m/z
实测
|/m/z
理论
*1000000《10ppm;且其实验二级质谱和相邻代谢物的拟二级质谱的相似度≥0.5。在实验数据中搜索与相邻代谢物m/z
理论
,t
r预测
以及拟二级质谱匹配的代谢物峰,匹配成功则该代谢物峰鉴定完成。如图2左下图所示,种子代谢物的二级谱图为下方红色谱图,将其作为4个相邻代谢物的二级谱图,每个相邻代谢物分别在实验数据中寻找与其m/z
理论
,t
r预测
,以及拟二级质谱匹配的代谢物峰。相邻代谢物hmdb0032913,c02110,hmdb0059883和hmdb0041011的m/z
理论
,t
r预测
分别为m/z 195.1749,12.69min;m/z 285.2218,17.11min;m/z 193.1592,13.38min和m/z 153.1279,9.42min。在实验数据中搜索与相邻代谢物满足阈值要求的代谢物峰,如图2左下图所示,在实验数据中搜索到保留时间14.32min,m/z 195.1745的代谢物峰,与hmdb0032913的|t
r预测-t
r实测
|/t
r预测
*100%=12.84%,|m/z
理论-m/z
实测
|/m/z
理论
*1000000=2.05ppm,且该峰的实验二级谱图(蓝色谱图)与hmdb0032913的拟二级谱图(红色谱图)的相似度为0.80,则该代谢物峰被定性为二羟基-β-紫罗兰酮(hmdb0032913)。为了进一步证实采用分子结构网络及相邻代谢物的“拟二级谱图”方法定性的可行性和可靠性,在图2右下图中对比了标准样品hmdb0032913的实验二级谱图(红色谱图)与混合标准样品中代谢物峰(14.32min,m/z 195.1745)的实验二级谱图(蓝色谱图),二者的二级谱图有很高的相似性,证明利用相邻代谢物拟二级谱图的分子结构关联网络定性方法是准确可靠的。采用上述定性方法,实验数据中2个代谢物峰(m/z
实测
,t
r实测
,二级相似度)m/z 193.1590,13.39min,0.96和m/z 153.1268,11.08min,0.54分别和α-紫罗兰酮(hmdb0059883)和β-环高柠檬醛(hmdb0041011)匹配,这2个代谢物峰也被成功鉴定。
[0033]
3)当实验数据搜索出多个匹配结果时,对匹配结果进行打分,得分规则为:
[0034]
得分=0.25*(1-(|m/z
理论-m/z
实测
|/m/z
理论
*1000000)/10) 0.25*(1-(|t
r预测-t
r实测
|/t
r预测
*100%)/30%) 0.5*二级谱图相似度
[0035]
其中,m/z
理论
为利用代谢物的分子式所获得的理论一级离子质荷比;m/z
实测
为实验测得的一级离子质荷比;t
r预测
为利用前述构建的保留时间预测模型所预测出的保留时间;t
r实测
为实验测得的代谢物峰的保留时间。
[0036]
如实验数据中搜索到3个代谢物峰满足搜索阈值,与hmdb0059883匹配,其m/z
实测
,t
r实测
,二级质谱相似度分别为m/z193.1581,13.38min,0.96;m/z 193.1583,13.49min,0.99和m/z 193.1590,13.39min,0.96,对3个结果进行打分,对应的得分为0.87,0.89和0.96,按照从大到小排序。得分越高的代谢物峰被鉴定的可靠性越高,但有多个搜索结果的代谢物峰不再作为新的种子参与下一轮定性。
[0037]
4)鉴定出的代谢物再作为新的种子,重复定性过程,直至没有新的代谢物被定性出来。如实验数据中代谢物峰被成功鉴定为二羟基-β-紫罗兰酮(图2中hmdb0032913),代谢物峰(m/z 195.1745,14.32min)的相应二级谱图被赋予下一级相邻代谢物hmdb0037139(二氢脱氢β-紫罗兰酮,dihydrodehydro-beta-ionone),作为其拟二级谱图。hmdb0037139的m/z
理论
,t
r预测
为193.1592,11.94min。在实验数据中寻找到满足阈值的代谢物峰m/z 193.1590,13.39min,其实验二级谱图与拟二级谱图相似度为0.72,匹配成功。代谢物峰(m/z 193.1590,13.39min)被成功鉴定为二氢脱氢β-紫罗兰酮,其作为新的种子重复上述定性过程。
[0038]
正离子模式下,885个代谢物混合标样的实验代谢物峰均符合一级质谱离子质荷比和保留时间的搜索阈值,从885个代谢物峰中选取60个代谢物作为种子代谢物映射到分子结构关联网络,采用上述鉴定过程,共可关联出578个代谢物峰,网络关联覆盖度为72.1%(638/885)。网络中未关联出的代谢物峰主要因为混合标样较少,在网络中缺少有效边连接。关联出的代谢物峰可满足全部鉴定阈值可成功鉴定的为461个代谢物峰(含种子),鉴定率为72.2%(461/638),其中392个代谢物峰鉴定结果正确,鉴定正确率为85.0%(392/461)。
[0039]
上述结果显示,本发明无需依赖于大规模实验二级谱图数据库的代谢组定性方法,可实现代谢组规模化快速定性,且结果准确可靠。
[0040]
实施例2
[0041]
为了进一步证实本发明在生物样本的提取物中同样有效可行,我们对动物组织(小鼠肝脏)进行了代谢组的提取,并对动物组织提取物进行超高效液相色谱-高分辨质谱数据采集,以采集的非靶向代谢组学数据的定性为例进行说明。
[0042]
组织代谢组的提取:采用动物代谢组学方法,提取小鼠肝脏中的代谢物。首先称取10毫克小鼠肝脏组织于1.5毫升离心管中,加入600微升的甲醇/水(体积比,4:1)提取剂,研磨珠打进行匀浆处理(25hz
×
1min*2),在4℃,12000rpm条件下离心15min。取400μl上清液,放入真空离心浓缩仪冻干。在冻干样品粉末中加入50微升乙腈/水(体积比,1:3),涡旋1min,在高速离心机中采用4摄氏度,14000rpm转速,离心10分钟。
[0043]
过程和条件同实施例1,与其不同之处在于:
[0044]
非靶向代谢组学数据采集:同实施例1。
[0045]
实验色谱-质谱信息获取:基于小鼠肝脏提取物的非靶向代谢组学数据,采用软件compounddiscovery3.1获得峰表,包括实验保留时间t
r实测
,一级质谱信息即一级离子质荷比m/z
实测
,导出excel表。采用软件proteowizard将原始数据进行转化,获得.mgf的二级文件,包含相应的二级质谱信息即二级离子的质荷比和强度。
[0046]
保留时间预测模型构建和分子结构关联网络构建过程同实例1所示,这里不再赘述。
[0047]
基于分子结构关联网络定性:以构建的分子结构关联网络作为背景网络,对非靶向超高效液相色谱-高分辨质谱代谢组采集的实验数据进行鉴定,确定待测生物样本中的代谢物。正离子模式下,512个代谢物峰在分子结构关联网络中符合一级质谱离子质荷比和保留时间的搜索阈值。采用同实施例1相同的鉴定过程,利用60个代谢物作为种子代谢物,共关联到422个代谢物峰,正离子模式的关联覆盖率为94.1%,其中成功鉴定353个代谢物
峰;负离子模式下,342个代谢物峰在分子结构关联网络中。采用同实施例1相同的鉴定过程,利用55个代谢物作为种子代谢物,共关联出258个代谢物峰,负离子模式的关联覆盖率为91.5%,其中成功鉴定230个代谢物峰。见图3,正负离子模式下共成功鉴定出518个代谢物。
[0048]
将分子结构关联网络定性方法与传统的搜库鉴定方法比较,以小鼠肝脏为例,采用compound discoverer(cd)质谱数据库,基于一级离子质荷比和二级质谱信息共鉴定到139个代谢物。分子结构关联网络定性方法基于保留时间,一级离子质荷比和二级相似性共鉴定出518个代谢物。如图4,本方法鉴定到更多的代谢物,且对传统方法所鉴定的代谢物有较好的包容性。本方法充分利用代谢组数据库中丰富的代谢物一级离子质荷比及其结构信息,发展不依赖大规模ms/ms数据库的基于分子结构关联网络的规模化代谢组定性方法,突破保留时间和二级质谱信息缺乏造成的定性瓶颈。从而,达到大规模、准确定性的目的。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。