用于测定硅中痕量金属的方法
1.本发明涉及一种用于测定硅中金属杂质的方法。
2.多晶体硅(多晶硅)被用作生产单晶硅的起始材料,例如通过坩埚区域拉制(切克劳斯基法)或通过区域熔融(浮区法)。所述单晶硅可以用锯片切为晶片,并且经过多个进一步加工步骤,可用于半导体行业来制造电子元件(如晶体管、二极管、双极晶体管和mos晶体管)。这些加工的步骤一般包括故意并且局部受限的引入掺杂剂杂质掺杂单晶硅。这些掺杂剂包括,特别是,具有三价或五价价电子的原子—就是例如所述元素是元素周期表中第3和第5主族元素。由于晶界和晶格缺陷是引起多余电流路径的可能原因之一,因此用来制造这些半导体元件的先决条件是使用的硅呈现极高的纯度(没有外来的掺杂剂)并且是一个完美的单晶形式。关于多晶硅的各种应用,对于所述杂质不得超过预定规定。
3.原则上,杂质(如硼、磷和砷)在ppt范围(份/万亿,10-12
)可能已经改变所需的硅半导体的性能。除了掺杂原子,尤其是痕量金属杂质和非金属杂质也可能导致缺陷。这里示例包括非金属碳和氯,和金属杂质包括铁、铬、镍或铜。
4.为了确保所述杂质低于所述说明的规定,适当的灵敏分析方法是必要的。此外,对于常规杂质监测、实践方面比如分析持续时间和再现性是重要的。
5.金属杂质中,一个基本的差别存在于多晶硅表面的金属和多晶硅本体中的金属之间。下文涉及一种用于测定硅本体中金属杂质的方法。
6.一种已知的方法是仪器中子活化分析(inaa)。通过这种方法,多晶硅样品表面清洗蚀刻处理后在核反应堆中辐照。所述金属污染物就在这种环境下转化成放射性同位素。来自放射性核分解的所述特定元素的γ射线被测量并且可以分配给该元素。
7.inaa的优点是它对于大多数金属具有非常低的检测极限。对于fe、cr和ni这些极限通常小于或等于50pg/g[pptw]硅。对于其它金属杂质,如ca或ti,检测极限是500至2000pg/g。缺点是需要用作中子源的使用仪器(核反应堆或粒子加速器)成本及复杂性巨大。另一个缺点是随着时间的推移需要大量的测量交错,因为不同元素拥有不同的分解的时间。inaa的持续时间可能是2到3个月。尽管inaa具有高检测实力,不适合作为一种进程内的分析技术(如常规分析用来优化参数),因为其需要短的测量持续时间。另外,这种分析非常昂贵。
[0008]
另一种技术是溶解硅样品无需事先浓缩杂质。在这种情况下,所述样品完全溶于由浓硝酸(hno3)和氢氟酸(hf)组成的含水的蚀刻溶液中,所述基体被烟熏,并且所述痕量金属再溶解于稀酸中。所述样品然后可以用痕量分析方法分析,例如原子吸收光谱法(aas),电感耦合等离子质谱(icp-ms),和电感耦合等离子体发射光谱法(icp-oes)。
[0009]
该优势在于分析运行时间短,通常1-2天。因为所述方法不包括浓缩步骤,与inaa相比,其检测极限通常大100倍。
[0010]
另一项技术是从ep 0 349 117 a2中获知被称为“凝固尖端”法。这种情况下,在第一步中,通过区域熔融(区域拉制)将该棒熔体中所述硅棒金属杂质进行富集浓缩。在区域熔融最后,能量供应停止,所述熔体凝固。所述具有浓缩杂质的冷却区域与所述合成的单晶分离后完全溶解在酸溶液中。该溶液然后可以通过其中一种所述的痕量分析方法分析。
[0011]“凝固-尖端”法的一个优点是它的运行时间一般不到一个星期,其适合用于常规分析。缺点是所述熔体或尖端自然冷却,且由此形成的所述结晶状态未定。对于这种情况的一个特定原因是这种方法仅仅是部分偏析,它会导致凝固的硅中所述金属污染物的不均匀分布。另一个缺点是所述冷却尖端需要从所述单晶上机械分离。这需要通过使用例如金刚石锯、金刚石刀具或舌头,通常此情况下导致的金属污染,必须在溶解之前消除。这种消除借助表面蚀刻进行,虽然不可避免的待分析的尖端部分也从实际的痕量金属分析中被去除和失去。有鉴于此分析结果可能会失真。
[0012]
de 10 2010 039 755 a1同样描述了一个基于区域熔融的方法,其和“凝固-尖端”法相比,通过特定的提拉技术在所述合成单晶和所述硅棒之间导致一个滴状的凝固,结晶样品部分(“凝固-凸起”)在所述单晶的端部。所述方法的一个优点是所述凸起不需要从所述单晶上机械分离,从而减少样品材料的污染和损失。缺点是相对低的回收率,总计大约10-40%。该原因是熔体中大部分的硅,在分离时其仍然保留在样品部分。这需要相对较高的检测极限。检测极限是由下列公式计算:
[0013]
其中
[0014]
dl:检测极限,
[0015]
rdv:重新溶解体积,
[0016]
rr:回收率,
[0017]msi
:重熔硅的质量
[0018]
σ:空白标准偏差值
[0019]
由于所述回收率在公式的分母中,低的回收率对应高的检测极限。此外,所述凝固凸起完全溶解需要相对大量的酸。这也导致更高的检测极限,因为所述酸含有杂质。
[0020]
本发明的目的是提供用于测定硅纯度的一种改进方法。
[0021]
该目的是通过测定硅中金属杂质(痕量金属和痕量半金属)的方法来实现,包括以下步骤:
[0022]
a)在区域熔融装置中准备一个棒状硅样品和一个棒状晶种;
[0023]
b)区域熔融(区域拉制)形成的单晶硅具有圆锥末端区域,在分离步骤中在单晶硅的末端形成滴状熔体;
[0024]
c)冷却所述滴状熔体以形成固化的硅滴(销凸起);
[0025]
d)在酸溶液中部分或完全溶解所述硅滴;
[0026]
e)通过痕量分析分析技术分析所述步骤d)中获得的溶液。
[0027]
该方法的特征在于,所述方法步骤b)中分离步骤包括以下顺序的子步骤:
[0028]-重熔硅样品以减少其直径,其在第一时间间隔硅样品和晶种的运动方向相对先前的方向是相反以形成所述圆锥形末端;
[0029]-形成滴状熔融区域,其中对于第二时间间隔所述晶种的运动停止并且所述硅样品的运动方向再次反转;
[0030]-晶种和硅样品的分离,其中所述硅样品的运动方向反转,所述样品的以150-400mm/min的速度持续运转5-20s。
[0031]
本发明的方法特别涉及de 10 2010 039 755 a1中所描述方法的向前发展和进一步改进。
[0032]
所述硅可以是单晶,多晶和多晶硅。优选多晶硅,更优选通过西门子法制备的多晶硅。
[0033]
从现有技术熟悉区域熔融装置中的区域熔融(也称为区域拉制或浮区(fz)法)。本文可参考特别是综述性文章硅区域拉制发展历史综述(w.zulehner-materials science and engineering:b,3april 2000,pp.7-15,elsevier)和de 10 2010 039 755 a1。
[0034]
简要概述,棒状硅样品(钻芯)通常是首先通过钻芯取样获取。此芯更具体涉及根据西门子法制备的多晶硅棒。所述硅样品通常有3-25cm的长度,优选是5-15cm,且直径是10-50mm,优选是15-25mm,更优选是17-21mm。
[0035]
被夹到区域熔融装置之前,所述硅样品通常被清洗(用溶剂和/或酸处理)。在使用区域熔融的情况下,所述硅样品的一端由感应线圈在一个狭窄的区域内熔融(感应加热)。所述熔融区(熔体)与棒状单晶硅接触,并在该棒状单晶硅上生长,获得该晶体的结构。将此熔融区然后慢慢移动穿过棒状硅样品。换句话说,两棒的运动方向被控制以使熔融区(感应线圈)沿着硅样品移动。在这个过程中,因此两个棒的运动的方向是相同的(参考图1)。由于所述硅样品和不断增长的单晶不同的运动速度,所述单晶的圆锥形末端区域被维持。所述两个棒旋转,从而使得所述区域熔体均匀。
[0036]
基于其圆柱区域,作为硅样品直径的函数,通常合成单晶的直径5-50mm,优选8-25mm,更优选10-15mm。所述晶体的长度通常依赖于所述硅样品的直径和长度。
[0037]
已经表明,与根据专利de 10 2010 039 755 a1所述的凝固-凸起方法相比,如果在滴状熔融区形成前再熔融使硅样品末端区域的直径减少,所述回收率上升。
[0038]
在使用凝固-凸起法情况下,在分离的时,所述硅样品和单晶之间有一个滴状熔融区。分离时,所述滴状熔融区主要部分(约50%-80%)在硅样品上再结晶,只有20%-50%在单晶硅上面再结晶形成所述凸起。所述硅样品上再结晶的部分在所述分析时损失。为降低该部分,本发明所述方法减小了所述硅样品末端区域的直径。也就是说,所述硅样品与所述滴状熔融区的接触区域直径减小。为了达到这个目的,与上述正常区域熔融相比所述单晶和硅样品的运动方向相反。持续一定时间段,因此,所述硅样品移动远离所述滴状熔融区或者所述感应线圈,导致硅样品的末端区域直径减少。
[0039]
这种方法的结果已显现意外惊喜,一小部分固化硅滴(凸起)中回收得到的所述金属杂质高达95%。相比之下,所述凝固-凸起法的平均回收率仅为40%左右。
[0040]
重熔后,所述硅样品长度1的末端区域优选具有一直径小于或等于所述单晶与所述熔体接触面的直径。
[0041]
所述单晶与所述熔体接触面的直径优选为3-8mm,更优选为4-6mm。所述硅样品端部长度l的直径优选为2-8mm,更有选是3-6mm。特别是,所述单晶接触面积的直径和硅样品长度l的末端区域直径约5mm(
±
0.5mm)。
[0042]
所述硅样品末端区域的长度l优选是其对应于直径的一到三倍。
[0043]
在重熔过程中,所述硅样品优选比所述单晶更高的速度移动。所述单晶的运动速度优选是对应硅样品运动速度的一半左右。
[0044]
所述硅样品的运动速度可能是5-15mm/min,优选7-13mm/min,更有选9-11mm/min。
[0045]
所述单晶的运动速度可能是2-10mm/min,优选3-8mm/min,更有选4-6mm/min。
[0046]
所述重熔期间第一时间间隔,其中有一方向反向的运转,优选是30-300s,更优选90-240s,最优选90-120s。
[0047]
所述重熔后,滴状熔融区形成。这是通过暂停晶种运转并且同步反转硅样品的运动方向来实现的。所述滴状熔融区在第二时间间隔形成,第二时间间隔持续时间优选1-4s,更优选2-3s。这里所述硅样品的运转速度可以是1-5mm/min,优选2-4mm/min。
[0048]
所述晶种和硅样品缓慢分离持续时间在5-20s,硅样品的运动速度是150-400mm/min,优选250-350mm/min,与所述滴状熔融区形成相比其运动方向再次反转的,并且单晶继续保持静止(除了旋转)。所述的感应加热最好在分离开始时的关掉。在分离后,所述滴状熔融区开始冷却。
[0049]
已经显现出来由于缓慢分离,本发明所有的杂质已经收集到固化硅滴的外层(销凸起),因此可能不需要完全溶解销凸起。
[0050]
关于所述浓缩的金属污染物发现于边缘区域外部的可能解释可能是吸气效应(参考w.zulehner-materials science and engineering:b,3april 2000,pp.7-15,elsevier).这里假设,当所述固体冷却,金属杂质在经受高温下溶解在固体中优先沉淀在缺陷部位,并因此积累在这些缺陷部位处。所述销凸起的表面,其晶格自然终止,因此在表面上具有开放的化学键,就是这样一个缺损部位。金属杂质的扩散速度通常是温度的函数,并且所述扩散在高温下比在低温下快(参考k.graff,硅器件制造中的金属杂质metal impurities in silicon-device fabrication,isbn 978-3-642-62965-5)。所述销凸起的缓慢冷却阶段可能因此增强吸气效应(所述硅样品的静止热熔融端缓慢的向前移动),所述金属杂质扩散到所述硅表面的自然缺陷部位。
[0051]
在步骤c)中的冷却,也可能停止所述硅样品的运动,同时让所述单晶按照原来的运动方向(区域熔融常用的运动方向)以150-400mm/min,优选250-350mm/min的移动速度运动。这将导致额外增加的所述吸气效应。
[0052]
然而,一般来说,步骤c)中的冷却还可以通过简单地选择关闭样品旋转让其停止下来实现。
[0053]
所述固化的硅滴(销凸起)通常具有的直径是3-10mm,优选6-8mm。所述销凸起通常对应低于1wt%的所述熔融硅样品。所述销凸起的重量(在没有附接到单晶硅)通常是3-5g。
[0054]
使用所述凝固凸起方法的情况下,没有必要把销凸起与所述单晶分开再溶解。冷却后,优选和该单晶一起转移到无尘袋里。它因此可以是非接触式夹持以及由此没有污染进入固定装置,并传送到湿化学消解程序。后者优选在洁净室条件下进行,在清洁室中合适的地方去除所述袋子。
[0055]
由于金属杂质主要位于所述销凸起的外层,不需要特别有利使所述销凸起完全溶解。根据浸渍持续时间,仅或多或少使厚外层溶解足以代替地将所述销凸起暂时沉浸在蚀刻溶液(酸)进行初步蚀刻。已经发现销凸起重约3g,所有的金属杂质包含于0.1-0.2g的脱硅。
[0056]
所述销凸起浸入酸之前,例如可任选地用稀硝酸除去游离酸进行表面清洁。然而,已发现这个过程可能的结果是污染的样品。优选直接传递至无尘袋。
[0057]
所述销凸起的浸酸持续时间优选为3-15min,更优选5-10min,最优选约6min。相比
于此,销凸起完全溶解的刻蚀时间平均为2小时。减短的蚀刻时间通常意味着更好的空白值,因此改进检测极限值。所述样品和酸暴露在周围的环境中的时间越长,它们将受污染的可能性就越大。
[0058]
所述酸或蚀刻溶液优选含有浓硝酸(50-80wt%)和氢氟酸(20-50wt%)的混合物,其比例为4:1至3:1,优选是2:1至1:1。所述蚀刻溶液可以在室温或加热(例如60℃)条件下使用。优选不进行加热。转移到一个加热装置构成又一污染源。
[0059]
一般而言,选择酸的总量刚好足够溶解销凸起的最外层。通常使用蚀刻液的总量是5-10ml。为了分离所述杂质的检测极限免遭化学物质的影响,化学制品的用量原则上应该最小化。所述销-凸起方法在这方面是优于凝固-凸起的方法。
[0060]
所述销凸起的部分溶解优选如下实现:
[0061]
1.提供所述蚀刻液,
[0062]
2.将所述销凸起浸没在蚀刻液中3-15min,
[0063]
3.用新鲜蚀刻溶液清洗从所述蚀刻液中移走的所述销凸起。
[0064]
4.通过加热蚀刻溶液到100-350℃来浓缩所述蚀刻溶液。
[0065]
随后根据使用的痕量分析测量技术,浓缩处理后的所述痕量金属剩余再溶解在稀硝酸(0.5-5wt%)和/或稀氢氟酸中得到测量溶液。由于减少了蚀刻液的去除并因此减少硅基质(溶解的硅以六氟硅酸的形式存在于蚀刻溶液中),与凝固-凸起方法相比,所述测量溶液中再溶解体积能够平均减少一半(通常从3到1.5ml)从而提高了金属杂质的浓度。因此测量可以在更大的浓度下进行,导致测量结果更稳定,因为该信号相对于噪声增强。
[0066]
对于步骤e)的分析,可能使用常用的质谱仪例如icp-ms。其它优选测量技术是电热原子吸收光谱法(已知石墨炉原子吸收光谱法、gfaas)和全反射x射线荧光分析(trfa)。
[0067]
图1显示了根据现有技术分离步骤之前的区域熔融。
[0068]
图2显示了销-凸起和凝固-凸起方法的比较。
[0069]
图3显示了单晶上的销-凸起。
[0070]
图4显示销-凸起在酸浴液中。
[0071]
图5示出了在蚀刻步骤中铁的浓度。
[0072]
图6示出了在蚀刻步骤中铜的浓度。
[0073]
图1示意显示了区域熔融装置10具有硅样品12和单晶14,其在分离步骤之前短暂存在。所述硅样品12和单晶14都被夹到一个旋转轴上,为清楚起见而未示出。所述转动方向如箭头11a,11b所示,所述运动方向如箭头13a,13b所示。所述硅样品12和单晶14通过熔融区域20彼此结合,该熔融区域被感应线圈21加热。所述硅样品12是一个棒状多晶硅样品具有一个圆柱形区域16和熔融区域20中的圆锥区域18。所述多晶硅样品的初始长度约为15cm。所述区域16的直径大约为20mm。所述单晶14在单个晶种15上再结晶由一初始圆锥体22,一个圆柱段24和熔融区域20中一圆锥部26组成。所述单晶14的长度大约110mm,圆柱段24的直径约14mm。
[0074]
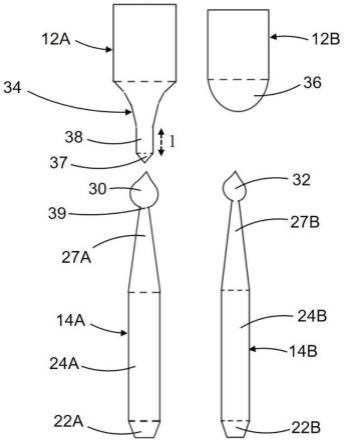
图2显示了根据本发明的方法生产的单晶14a,在其圆锥形末端区域27a有一个销凸起30。如图所示,与本发明相比的是凝固凸起32(根据de10 2010 039 755a1),其在单晶14b的一个圆锥形末端区域27b上。所述末端区域27a,27b可具有的长度是3-6cm。所述部件的编号已在图1中标记的仍然保留,源自销凸起方法得到的部件通过一个“a”区别并且源自
凝固凸起方法得到的部件通过一个“b”区别。也示出了用所述方法获得相应的硅样品12a和12b,及它们末端区域34和36。所述单晶24a,24b在形状上没有实质性差异。所述圆柱段24a,24b的直径没有任何必然差别。这两个方法实施后基本可见的区别在于所述单晶12a、12b的末端区域34,36。用凝固-凸起方法实施的情况下,处于熔体部分的所述硅样品12b在分离期间形成一个圆头的末端区域36。此末端区域36大体上相应于50-80%的熔体。因此仅20-50%保留在所述单晶24b的圆锥形末端区域27b的凝固凸起32。相反,对于销-凸起的方法,由于重熔,末端区域34与具有长度i的部分区间38一起形成,其就直径而言至多对应于圆锥形末端区域27a和销-凸起30之间的接触面39。按照这种方式,所述硅样品12a的一部分在分离期间处于所述熔体减小为很小部分37。这部分37通常来说对应于仅5-10%的分离过程中所述熔体。通常,所以所述分析只有5-10%的硅熔体损失。
[0075]
图3显示了冷却后在所述单晶的14a细的锥形末端区域27a形成销-凸起30(参考图2)。标注的外层40是想要示范大约的所述蚀刻去除量,这为后续的分析提供硅使其部分溶解在酸中。所述金属杂质被包含在外层40内。与所述销-凸起30接触的接触面39的直径约5mm。这个直径也基本上对应熔体分离时所述单晶14a与锥形末端区域27a的接触面直径。
[0076]
图4示出了在底座52中有一装有酸的圆锥形容槽50。所述单晶14a具有的销凸起被浸渍在所述容槽50,其通过固定支架54固定。由于所述圆锥形容槽,所述用来部分溶解的酸的量可以减少。
[0077]
实施例1
[0078]
选自用切克劳斯基方法拉制的硅棒的6钻芯(硅样品)都具有22mm的直径,约8cm的长度并被已知的污染物金属fe,cr,ni,cu,zn和sn污染。在hf(45%)和hno3(65%)以1:6配比的混合酸中清洗蚀刻所述样品约15min,后用超纯净水漂洗。为此目的,制备需洁净室条件下(10级)进行。所述硅样品每个安装到fz装置(上部拉轴:硅样品;下部拉轴:晶种)。所述晶种可采用单晶硅。所述区域熔融操作在表1中陈述,也可参考附图。用负号标记的运动速度v表示当熔融区穿过所述硅样品时通常的拉力方向。参照图1和2,负的运动速度表示所述单晶14a和/或硅样品12相对于感应线圈21向下运动。正号表示向上的移动。
[0079]
[0080][0081]
表1
[0082]
单晶14a的的旋转速度通常为15-30rpm,并且相反方向旋转硅样品12a转速通常为3-10rpm(参考图1,箭头11a,11b)。分离和冷却后,销-凸起的最大直径约8mm。
[0083]
拆下后,在洁净室条件下(10级)将销-凸起30用固定支架54转移到一个装有酸的全氟烷氧基烷烃(pfa)锥形容器50使其部分溶解(部分蚀刻)。所述酸由hno3(69wt%)和hf(40wt%)按比例1:1的混合物组成。该容器50中装有约6ml的酸。所述完全浸没的销-凸起被暴露于所述酸中6min。该销-凸起随后用1ml新鲜的酸洗涤。将所得蚀刻后溶液然后在250
°
的温度下浓缩约30min。使用25μl的hf(40wt%)、25μl的hno3(65wt%)和1450ml超纯水的混合液溶解得到的残留物得到测量溶液。所述六个销-凸起各自用该处理获得。
[0084]
通过icp-ms方法进行测量,测定元素fe、cr、ni、na、k、sn、zn、al、cu、mo、la、cs、ce、
te、sc、se、ti、ta、ge、w、mg、ag、li、v、mn、zr、pb、y、sr、ba、bi、cd、sn、as、ru、rb、u、ga、in、ca和co。同样地生成空白值。为了实现这个目的,4个锥形容槽被填充酸并且不含样品按照类似的方式进行处理。在这些实施例的环境中这些空白值被测定取平均值,并在每次测量时从所述样品的测定量中减去。然后计算的值是表示相对于所述的单晶的总重量。
[0085]
这表明与凝固-凸起方法相比对于所选金属具有高得多的回收率(rr)。
[0086]
所考虑的值是所有六个硅样品是平均值。
[0087][0088]
表2
[0089]
表3显示与所述凝固-凸起方法相比减少了检测极限。
[0090]
dl(pg/g)fecrnicuznna凝固-凸起≤20≤20≤20≤10≤10≤20销-凸起≤2≤1≤1≤1≤1≤1
[0091]
表3
[0092]
图5和6显示了销-凸起在经过5min、16min、29min、44min和79min部分蚀刻后获得的测定溶液中所述铁的浓度曲线图(c
fe
,图5)和铜的浓度曲线图(ccu,图6)(蚀刻步骤:每个步骤之后,所述销-凸起被转移到新鲜的蚀刻液)。制备铬和镍相应的曲线,每个实验重复一次(样品1和2对于每种情况)。结果显示在表4中。
[0093][0094]
表4
[0095]
在所有情况下,蚀刻仅5min后,去除仅为0.06g,至少96%的所述金属杂质已蚀刻
掉。该结果表明通过清洗凝固尖端(参考ep 0 349 117a2)用清洗蚀刻液,用来分析的至少一部分金属杂质已经丢失。
[0096]
实施例2
[0097]
在区域熔融中测试不同的参数
[0098]
使用19和22mm的7硅样品(钻芯)。通过调整区域熔融相应的参数,获得不同直径(d)的所述单晶和所述硅样品的末端区域。此外,再熔期间所述下部拉轴的不同移动长度被试验,并获得了不同尺寸的销-凸起。此外,不同重量的单晶样品被拉伸和测试。结果如表5所示。
[0099][0100]
表5
[0101]
回收率始终介于90%和97%之间。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。