1.本发明涉及一种生物材料的制备方法,由该方法获得的生物材料,以及该生物材料作为组织工程支撑物、用于细胞培养或扩增、作为可植入医疗装置和作为药物的用途。
2.因此,本发明在医学领域具有实用性,特别是用于组织工程、药物递送、伤口敷料和植入物。
3.在下面的描述中,对括号([])的引用是指位于文本末尾的参考文献列表。
背景技术:
[0004]
生物材料广泛用于各种治疗应用,例如组织工程、药物输送、伤口敷料和植入物。
[0005]
合成的、活的或混合的,它们在几十年内获得了所有治疗领域:心血管、外科和骨科、牙科、眼科、皮肤科、泌尿科、肾科、神经科、内分泌科,尤其是再生或改善组织的功能。
[0006]
有几种类型的生物材料,但可以设想四种主要类别的生物材料:
[0007]-金属和金属合金,
[0008]-广义上的陶瓷,
[0009]-聚合物和软物质,
[0010]-天然来源的材料,例如珊瑚或从植物或动物有机体中提取的其他成分,例如甲壳素、藻酸盐、肝素、褐藻糖胶、纤维素、胶原蛋白或纤维蛋白。
[0011]
最后一类生物材料特别有趣,因为天然来源的材料通常具有天然的生物相容性和可生物降解性。
[0012]
涉及热聚集或交联的多种技术已用于制备基于天然材料的生物材料。然而,使用高温和交联剂可能导致材料结构发生不可逆的变性和改变,导致其生物学特性丧失,并可能揭示新的抗原位点,从而引发炎症反应。
[0013]
因此,需要基于不具有这些缺点并且满足更多天然生物材料需求的天然来源材料的替代生物材料。
技术实现要素:
[0014]
通过广泛的研究,申请人已经开发出新的生物材料,其在水、酸性溶液和细胞培养基中表现出显著的机械性能和良好的稳定性。
[0015]
令人惊讶的是,获得的生物材料表现出类似固体的行为,使其成为组织工程或细胞培养的支撑物的良好候选物。
[0016]
本公开报道了可能完全由一种独特类型的蛋白质或几种在生物材料制备过程中变性/未变性的选定蛋白质制成的第一生物材料。这意味着本发明的生物材料不太可能在受试者中引发炎症反应。此外,生物材料无细胞毒性,并且有利于细胞粘附和定植。
[0017]
由于它是由蛋白质制成的,本发明的生物材料是完全可生物降解的。
[0018]
申请人开发的制备方法可以在没有任何化学或苛刻/变性剂配制条件的情况下获得生物材料。它还允许调节生物材料的特性,特别是机械和内在特性,并获得具有新特性的
功能化生物材料。因此,申请人提供了可以调节其特性以适应靶向治疗应用的要求的通用生物材料。
[0019]
因此,在第一方面,本发明提供了一种制备生物材料的方法,包括以下步骤:
[0020]
a)制备包含至少一种在水中的溶解度高于或等于约10mg/ml的蛋白质和至少一种在水中的溶解度高于或等于约500mg/ml的盐的溶液,
[0021]
b)在4至50℃的温度下在大气压下或在更低温度下在真空或低于大气压的压力下,将步骤a)中获得的溶液原样蒸发,作为通过使步骤a)中获得的溶液发泡而获得的泡沫,或其混合物,直到形成两个不混溶相或直到获得基本上干燥的固体,
[0022]
从而获得生物材料。
[0023]
本发明的方法具有不使用任何共价交联或热聚集步骤的显著优势,从而允许获得具有非变性蛋白的生物材料。
[0024]
步骤a)中使用的至少一种蛋白质可以是在20℃的温度下在水中的溶解度高于或等于约10mg/ml,例如高于或等于20mg/ml、或40mg/ml、或50mg/ml的任何蛋白质。例如,蛋白质可具有介于约10mg/ml和1000mg/ml之间的溶解度。蛋白质在水中的溶解度可以通过本领域技术人员已知的任何方法测量,例如高效液相色谱(hplc)、衰减全反射(atr)-ftir光谱、拉曼光谱或聚焦光束反射模式(fbrm)测量。此类蛋白质可以例如选自血清蛋白质,例如白蛋白或球蛋白,尤其是γ-球蛋白。白蛋白可特别选自人血清白蛋白、牛血清白蛋白、猪血清白蛋白、卵清蛋白、植物白蛋白和重组白蛋白,例如在结构上与天然人血清白蛋白等效并在稻米中产生的重组人白蛋白。白蛋白也可以是白蛋白纳米颗粒。如上所述,“至少一种”蛋白质可用于本发明的方法中,意味着1、或2、或3、或4种或更多种不同的蛋白质可用于步骤a)的溶液中。优选地,它可以使用1到3种不同的蛋白质。步骤a)的溶液中蛋白质的浓度可以是允许与盐混合的任何浓度。它可以例如包含在10mg/ml和500mg/ml之间。本领域技术人员能够根据盐的性质和盐浓度调整溶液中蛋白质的浓度。然而,如下文更详细解释的,盐和蛋白质之间的摩尔比比蛋白质浓度更重要,以获得具有期望特征的生物材料。
[0025]
步骤a)中的至少一种盐可以是在20℃的温度下在水中的溶解度高于或等于约500mg/ml,例如高于或等于700mg/ml、或900mg/ml、或1200mg/ml的任何盐。例如,盐的溶解度可以在约500mg/ml和3000mg/ml之间。该盐在水中的溶解度可以在约20℃的温度、ph 7、大气压下测量。盐在水中的溶解度可以例如通过本领域技术人员已知的任何方法来测量,例如离子色谱法、气相色谱法、酸碱滴定法、电位滴定法、容量法、称重法或拉曼光谱法。至少一种盐可以例如选自nabr、nai、ki、cacl2、mgcl2、kc2h3o2和nh4hco2。如上所述,“至少一种”盐可以用于本发明的方法中,这意味着可以使用1种、或2种、或3种、或4种或更多种不同的盐。优选地,它可以使用1-3种盐。例如,所述至少一种盐可以包含nabr和nai、nabr和cacl2、ki和cacl2或nabr/cacl2/mgcl2,例如摩尔比为100/100/100、200/200/200或300/300/300。步骤a)的溶液中盐的浓度可以是允许与蛋白质混合的任何浓度。它可以例如在0.01m和40m之间。本领域技术人员能够根据蛋白质的性质和蛋白质浓度来调整溶液中盐的浓度。然而,如下文更详细解释的,盐和蛋白质之间的摩尔比比盐浓度更重要,以获得具有期望特征的生物材料。
[0026]
优选地,在步骤a)中,至少一种蛋白质和至少一种盐的摩尔比取决于蛋白质的性质和用于获得生物材料的盐的性质。由于申请人已经证明生物材料的形成取决于蛋白质和
盐浓度的配对效应,因此盐/蛋白质的摩尔比是评估膜形成的更相关和更可靠的参数。知道这点,摩尔比可由技术人员根据其一般知识和不溶性生物材料的期望性质,特别是其硬度来确定,而没有过度负担。例如,摩尔比可以包括在100和4000之间,例如在100和3000之间,或在300和2500之间,或在400和2000之间,或在600和1500之间,或在650和1000之间,这取决于步骤a)中使用的盐和蛋白质。例如,nabr和白蛋白的混合物的摩尔比可以是664。
[0027]
步骤a)的溶液可以通过在非变性条件下将至少一种蛋白质和至少一种盐在适合的溶剂或溶剂混合物中混合来实现。技术人员可以根据其一般知识以及盐和蛋白质的性质来选择溶剂,而没有过度的负担。例如,溶剂可以选自水、缓冲液如乙酸盐缓冲液、水和缓冲液以及和另一种水混溶性溶剂如乙醇、甲醇、丙酮、dmf或dmso的混合物。在步骤a)期间溶液的温度可以在5℃和40℃之间。
[0028]
步骤a)可以在避免蛋白质变性的任何ph下进行,这是本领域技术人员已知的。优选地,步骤a)在包括在3.0和9.0之间的ph下进行,3.0的值被任选地排除。ph可以例如在4.0和9.0之间,或在4.0和8.0之间。
[0029]
混合物可以实现或可以转移到适合接收这种混合物的任何容器或支撑物上。例如,它可以是玻璃或硅树脂的模具、显微镜或微阵列基板、细胞和组织培养皿或微孔板,或生物材料可以围绕其成形的聚合物支撑物。有利地,可以根据要获得的生物材料的期望表面积、形状和厚度来选择支撑物。为此目的,待倒入容器中的混合物的体积可以根据支撑物的表面积和/或生物材料的期望厚度来选择。生物材料可以具有任何形状,例如膜、实心或空心圆柱体、圆锥体、球体、路面。作为信息,比率m/s是用于制剂的蛋白质的初始重量与如下例示的容器的面积之间的比率,可以包括在10mg/cm2和400mg/cm2之间,用于例如在20mg/cm2和400mg/cm2之间。
[0030]
步骤b)的蒸发可以对步骤a)中获得的溶液原样进行。在这种情况下,步骤b)直接在步骤a)之后进行,或者在不改变步骤a)中得到的溶液的性质或物理结构的中间步骤之后进行。
[0031]
或者,步骤b)可以对通过使步骤a)中获得的溶液起泡而获得的泡沫进行。例如,可以通过对步骤a)中获得的溶液施加机械功以增加溶液的表面积来获得泡沫。这可以通过本领域技术人员已知的任何方法进行,例如搅拌、将大量气体分散到步骤a)获得的溶液中,或将气体注入步骤a)获得的溶液中。
[0032]
在另一个实施方案中,步骤b)可以对步骤a)中获得的溶液和通过使步骤a)中获得的溶液起泡而获得的泡沫的混合物进行。在这种情况下,可以将步骤a)中获得的部分溶液取出并在单独的容器中发泡。所得泡沫可按原样蒸发或与溶液一起放回并与其混合,然后如步骤b)中蒸发。优选地,轻轻地进行混合以保存泡沫。泡沫的蒸发产生高度多孔的生物材料。
[0033]
进行步骤b)的蒸发以允许形成两个不混溶相或获得基本上干燥的固体。有利地,在可以避免存在于溶液、泡沫或其混合物中的蛋白质变性的条件下进行。为此目的,可以确定温度和压力,并相对于彼此进行调整,以实现该目标。本领域技术人员能够根据蛋白质或盐的种类并根据其一般知识来确定这些参数。例如,温度可包括在大气压下从4℃至50℃,例如从4℃至20℃,或从10℃至50℃,或从15℃至50℃,或从20℃至50℃,或25至40℃,或25至35℃,或20至30℃。还可以在真空下或在低于大气压的压力下在较低温度下进行步骤b)。
在这种情况下,温度可以为例如1℃至20℃,或2℃至15℃,或5℃至10℃,压力可以为1至100kpa。在任何情况下,进行蒸发,直至形成两个不混溶相或直至获得基本上干燥的固体。例如,当薄层盐沉积在生物材料的表面上时,或者当获得包含按重量计少于20%的水,例如少于10%的水的固体时,执行此操作。两个不混溶相或基本上干燥的固体的形成是视觉可识别的。有关信息,可以任选地通过重量分析测量水分来验证。
[0034]
蒸发阶段的持续时间可由本领域技术人员根据其一般知识确定而没有过度负担。这可能取决于蛋白质的种类、盐的种类、选择用于执行该过程的温度和压力、要蒸发的溶液的体积或容器的形状。例如,蒸发可进行10小时至30天,例如1天至20天,或2天至30天。可以执行更长的持续时间,但通常不会对生物材料的技术特征进行任何改进。
[0035]
步骤b)可以在避免蛋白质变性的任何ph下进行,这是本领域技术人员已知的。例如,步骤b)可以在包括在3.0和9.0之间的ph下进行,3.0的值被任选地排除。取决于蛋白质的种类,ph可以例如在4.0和9.0之间,或在4.0和8.0之间。
[0036]
蒸发可以通过满足上述标准的任何方式进行,例如烘箱或真空烘箱。
[0037]
本发明的方法可以由如上所述的步骤a)和b)组成,因为它们允许获得本发明的生物材料。在这种情况下,该方法没有任何其他步骤,并且可以在步骤b)结束时直接获得生物材料,因为它可以是基本上干燥的固体,或者是在步骤b)中获得的两个不混溶相的固相。
[0038]
或者,本发明的方法可以包括允许获得本发明的生物材料的附加步骤。附加步骤可以在步骤a)之前,和/或在步骤a)和b)之间,和/或在步骤b)之后进行。在这种情况下,可以在执行这些附加步骤之后获得生物材料。
[0039]
例如,可以将步骤b)中获得的固相或干燥固体洗涤,以除去至少一部分盐,从而获得生物材料。优选地,步骤b)中获得的固相或干燥固体可以被洗涤,直到至少90重量%的至少一种盐被消除,从而获得生物材料。可以进行洗涤,直至消除例如至少95%或至少99重量%的所述至少一种盐。洗涤可以通过技术人员已知的任何方式进行,例如用蒸馏水或含水缓冲液。所得盐浓度的控制可以用任何已知方法进行,例如通过如下所示的bca或微量分析。
[0040]
可以例如在洗涤之后进行步骤b)中获得的固相或干燥固体的浸泡步骤。浸泡可以使生物材料水合。浸泡可以通过本领域技术人员已知的任何方式进行,例如用蒸馏水或缓冲液,在室温下浸泡48小时。
[0041]
还可以在步骤a)和/或步骤b)期间,或在如上所述的任何附加步骤期间,添加至少一种添加剂。添加剂可以是允许根据需要调节生物材料特性的任何物质。在某些情况下,添加剂还可以允许从生物材料中去除盐或其一部分。添加剂可以由技术人员根据其一般知识和生物材料的期望特性来选择。它们可以例如选自聚合物,特别是不带电的、带正电的、带负电的和两性离子聚合物、非离子氨基酸和颗粒。聚合物可以是选自多糖、蛋白质、肽和多核苷酸和/或合成和半合成聚合物的任何天然聚合物。例如,聚合物包括但不限于多肽、均聚肽、壳聚糖、透明质酸、肝素、海藻酸盐、硫酸软骨素、聚精氨酸、聚赖氨酸、ε-聚赖氨酸、deae葡聚糖、聚环糊精、聚烯丙胺盐酸盐、聚乙烯亚胺、黄原胶、聚丙烯酸、聚乙二醇、淀粉、纤维素及其衍生物、胶原蛋白、胰岛素、纤维蛋白原、酪蛋白、明胶、麦醇溶蛋白、谷蛋白、弹性蛋白、球蛋白和血红蛋白。氨基酸可以选自所有合适的氨基酸,优选地选自精氨酸、鸟氨酸、赖氨酸和半胱氨酸。颗粒可以是任何合适的颗粒,并且可以例如选自纳米颗粒,例如碳
纳米管或石墨烯、微粒、细菌和病毒载体。添加剂可以通过本领域技术人员已知的任何方式掺入。例如,可以通过将所述添加剂直接溶解和/或悬浮于步骤a)中获得的溶液中,或通过将所述添加剂溶解和/或悬浮于水混溶性溶剂中,然后将混合物掺入步骤a)中获得的溶液中,将添加剂加入到步骤a)中获得的溶液中。另外地或可替代地,添加剂可以通过将溶解和/或悬浮在溶剂中的所述添加剂吸附到步骤b)中获得的生物材料上而掺入步骤b)中获得的生物材料中。添加剂的量可以适合于蛋白质和生物材料的期望性质,因此可以由技术人员根据其一般知识来确定。例如,相对于生物材料中蛋白质的总量,添加剂的百分比可为0至20重量%,例如1至18重量%,或2至15重量%,或3至12重量%。
[0042]
另外地或替代地,可以将至少一种活性成分掺入步骤a)中获得的溶液和/或步骤b)中获得的生物材料中。这可以允许对生物材料进行功能化。可以通过技术人员已知的任何方式掺入活性成分。例如,可以通过将所述活性成分直接溶解和/或悬浮于步骤a)中获得的溶液中,或通过将所述活性成分溶解和/或悬浮于水混溶性溶剂中,然后将混合物掺入步骤a)中获得的溶液中,将活性成分加入到步骤a)中获得的溶液中。另外地或可替代地,活性成分可以通过将溶解和/或悬浮在溶剂中的所述活性成分吸附到步骤b)中获得的生物材料上而掺入步骤b)中获得的生物材料中。在这种情况下,溶剂可以选自水、有机溶剂或水和水混溶性溶剂的混合物。有利地,可以将至多30%(v/v)的有机溶剂添加到蛋白质/盐溶液中,而不会抑制膜形成或显著改变所制备材料的性质,从而允许它们用于掺入水不溶性活性成分。例如,取决于蛋白质和溶剂的性质,可以加入至多1%、或2%、或3%、或4%、或5%、或6%、或7%、或8%、或9%、或10%、或11%、或12%、或13%、或14%、或15%、或16%、或17%、或18%、或19%、或至20%、或21%、或22%、或23%、或24%、或25%、或26%、或27%、或28%、或29%或30%(v/v)的有机溶剂。它可以是例如至多15%乙醇、10%dmso或30%乙腈或二氯甲烷。本领域技术人员能够根据蛋白质、盐和溶剂的功能通过进行常规实验来调整该量。有利地,可以将至多30%(v/v)的有机溶剂添加到bsa/盐溶液中,而不会抑制膜形成或显著改变所制备材料的性质,从而允许它们用于掺入水不溶性活性物质。
[0043]
活性成分可以是允许赋予生物材料有趣特性的任何物质。活性成分可以由技术人员根据其一般知识和生物材料的期望特性来选择。它可以选自抗癌物质,例如戈舍瑞林、亮丙瑞林、卡莫司汀、紫杉醇、组氨曲林或吉西他滨,抗炎剂例如双氯芬酸,免疫抑制剂例如硫唑嘌呤或甲氨蝶呤,免疫调节剂例如环孢菌素,细胞外基质相互作用调节剂,包括细胞生长抑制剂如伊马替尼或阿西替尼,抗凝剂如利伐沙班或依度沙班,抗血栓剂(antithrombotic agent)如氯吡格雷,酶抑制剂,镇痛剂如吗啡或氢可酮,抗增殖剂,抗真菌物质(antimycotic substances),细胞生长抑制物质(cytostatic substances),生长因子如促红细胞生成素或血小板生成素,酶,激素,类固醇如氢化可的松或泼尼松龙,非类固醇物质和抗组胺药如苯海拉明或非索非那定,该列表不是限制性的。活性成分的量可以适合于蛋白质和生物材料的期望性质,因此可以由技术人员根据其一般知识来确定。例如,相对于生物材料中蛋白质的总量,活性成分的百分比可以为0至30重量%,例如0至25重量%,或1至25重量%,或1至20重量%,或1至18重量%,或2至15重量%,或3至12重量%。
[0044]
在任何情况下,通过本发明的方法获得的生物材料可以包含相对于生物材料的总重量的至少50重量%的蛋白质,例如至少55%、或至少60%、或至少70%、或至少80%、或至少90%、或甚至100%。
[0045]
通过实施该制备过程获得的生物材料是本发明的第二个目的。
[0046]
如上所述,生物材料的特性,包括其视觉外观,可以通过修改制备过程的一个或多个参数来调节,特别是盐的种类和盐/蛋白质的比例,从而提供根据需要调节生物材料的特性的可能性。
[0047]
关于生物材料的坚固性,它可能是固体、不溶性生物材料,范围从泡沫到致密材料,包括水凝胶。
[0048]
关于生物材料的形状,它可以是任何期望的形状,这取决于设想的应用和用于制备生物材料的容器。它可以是膜、管、圆柱体、垫、环,该列表不是限制性的。生物材料也可以在实施该过程之后被切割以获得期望的形状或尺寸。
[0049]
通过研磨生物材料,生物材料可以具有任何期望的尺寸,包括微粒。
[0050]
生物材料的视觉方面可以是半透明到不透明的。
[0051]
如上所述,本发明的生物材料的主要优点之一是用于制备生物材料的蛋白质在本发明的制备过程中不会变性。该制备在非变性条件下进行,并且可以在步骤a)中获得的溶液和步骤b)中获得的生物材料中分析蛋白质的形状和/或二级结构。根据本发明,“未变性”是指在步骤a)中获得的溶液中所述蛋白质的二级结构百分比至少与用类似浓度的相应天然蛋白质制备的对照溶液中相同或与在对照干燥天然蛋白质粉末中的相同,并且在步骤b)中获得的生物材料中所述蛋白质的二级结构百分比与相应的天然蛋白质相比存在至少显著增加的β-转角和分子间β-折叠以及显著减少的无序结构。生物材料中蛋白质二级结构的形状和百分比可以通过本领域技术人员已知的任何方法来控制,例如通过ir分析或saxs(小角x射线散射),如下文说明。
[0052]
本发明的生物材料随时间特别稳定。有利地,它在水溶液中、在酸性、中性和碱性ph中,和/或在有机溶剂如乙醇中稳定至少2天,优选至少7天期间。这意味着在此期间生物材料基本上没有溶解,即使可能观察到其结构因氢键和二硫键断裂而改变,并且质量损失低于20%。例如,在碱性溶液中质量损失可至多为20%,在水、乙醇和酸性溶液中至多10%。生物材料的质量损失可以如下面的实施例所说明计算。
[0053]
如上解释,生物材料可以仅由蛋白质制成,特别是非变性蛋白质,这意味着良好的生物相容性。有利地,生物材料可以与至少一种如前所述的活性成分结合以改善其生物学特性。
[0054]
因此,本发明的另一个目的涉及本发明的生物材料作为体外组织工程和/或体外细胞培养和扩增的支撑物和/或可植入医疗装置的用途。实际上,本发明的生物材料为细胞生长提供了必要的结构和生化支持,并且由于它们可能是三维的,它们特别适用于细胞培养和药物/细胞递送。
[0055]
本发明的另一个目的涉及该生物材料用作药物的用途。由于本发明的生物材料能够与生物系统相互作用,因此它可以例如用于构建用于诊断目的的装置、组织或器官替代物或功能替代的装置。因此,本发明的生物材料可在体内用作可植入医疗装置,特别是用于替换有需要的受试者的缺陷组织,或用于药物释放系统。换言之,本发明还描述了包含本发明的生物材料的可植入医疗装置。有利地,取决于生物材料的性质,包含本发明的生物材料的装置或药物可以在一段时间后被再吸收到活体中。例如,该时间段可以是在插入活体后20天之后,或30天,或超过60天。
[0056]
本发明通过以下关于附图的实施例进一步说明,附图不应被解释为限制性的。
附图说明
[0057]-图1表示在盐存在下通过蒸发制备基于白蛋白的生物材料。所选制剂的nabr/bsa(牛血清白蛋白)摩尔比为664。蒸发在大气压下的烘箱(37℃)中进行7天,直到生物材料完全干燥。过量的盐在生物材料表面形成薄层。蒸发后,采用洗涤和浸泡步骤去除盐,留下不溶于水的基于白蛋白的生物材料。
[0058]
●
图2表示随着时间对bsa/nabr 664膜的形成的跟踪(在37℃蒸发、ph=6、m/s=105mg/cm2)。洗涤后,膜是透明的。模具底部覆盖有不粘有机硅盘。右上角出现的不规则是由于气泡造成的。
[0059]
●
图3表示在增加浓度的nabr(ph=6)存在下白蛋白的表面电荷滴定。
[0060]
●
图4通过振荡方案(频率变化:100hz至0.01hz,控制的剪切应力:1pa)表示基于白蛋白的膜的流变特性。剪切模量(pa)的弹性分量用实线表示;剪切模量(pa)的粘性分量用虚线表示。
[0061]
●
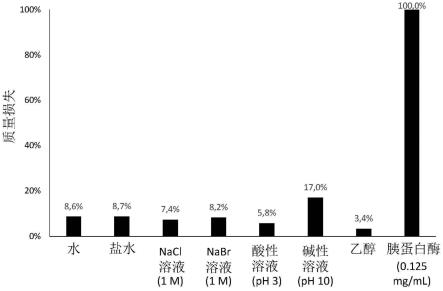
图5表示bsa/nabr 664膜(在37℃和ph=6下蒸发)在各种溶出介质中的稳定性(从左到右:水、盐水、nacl溶液1m、nabr溶液1m、酸性溶液ph 3、碱性溶液ph 10、乙醇和胰蛋白酶0.125mg/ml)。对于每种溶出介质,将膜置于25ml介质中。实验在37℃和搅拌下进行7天。
[0062]
●
图6表示在蒸馏水2-巯基乙醇溶液(2me,0.1m,)和尿素溶液(2m,4m和8m)中培养的bsa/nabr 664膜(在37℃和ph=6下蒸发)的流变特性。对于溶液,将一批2个膜置于30ml介质中。实验在室温下进行24小时。通过振荡方案(频率变化:100hz至0.01hz,控制的剪切应力:1pa)评估膜的流变特性。
[0063]
●
图7表示(a)bsa溶液(100mg/ml,在d2o中)中存在的白蛋白的酰胺i带中(amid i band)的拟合和每个二级结构的子带的识别(拟合曲线和原始的酰胺i带光谱重叠,残余rms误差《0.005)和(b-e)比较(b)bsa对照溶液(100mg/ml,d2o,实线)与bsa/nabr 664溶液(bsa:100mg/ml,nabr:1m,d2o,虚线),(c)bsa对照粉末(实线)与bsa/nabr 664膜(虚线),(d)bsa对照溶液(100mg/ml,d2o,实线)与bsa对照粉末(虚线)以及(e)bsa/nabr 664溶液(bsa:100mg/ml,nabr:1m,d2o,实线)与bsa/nabr 664膜(虚线)中bsa的酰胺i带。
[0064]
●
图8a和图8b表示图8a)bsa溶液(溶液2,40重量%h2o)的散射曲线和使用crysol计算的理论强度,使用了来自3v03 pbd原子坐标的单体bsa蛋白(50次谐波,排除体积8.7 104,其他参数为默认值)。在理论曲线上应用比例因子以匹配数据。图8b)(a)bsa干粉、(b,c)bsa溶液((b)溶液1含24.21重量%h2o,(c)溶液2含39.09重量%h2o)、(d)nabr干粉(*表示111、200和220布拉格反射)、(e)bsa/nabr 664膜的x射线散射曲线。为了清楚起见,将数据垂直移动。
[0065]
●
图9表示在选定条件下(在37℃和ph=6下蒸发),以664的摩尔比nabr/bsa配制的基于白蛋白的膜的sem分析。样品在观察前被金属化。a)膜表面;b)膜的横截面。
[0066]
●
图10表示用膜提取物(bsa/nabr 400、bsa/nabr 664和bsa/cacl
2 700,12.5%、25%、50%和100%)处理的balbc 3t3成纤维细胞的细胞活力。通过比较与bsa/nabr 400、bsa/nabr 664和bsa/cacl
2 700提取物接触24小时内培养的balbc 3t3细胞的归一化代谢
活性与阳性对照(ctl )的归一化代谢活性来估计间接细胞毒性。(*)在处理组和阳性对照(ctl )之间观察到显著差异(p《0.05)。生物复制数=1,总技术复制数=4。
[0067]
●
图11表示与白蛋白膜直接接触培养的balbc 3t3成纤维细胞的细胞活力。通过比较与bsa/nabr 400bsa/nabr 664和bsa/cacl2(m/s=25mg/cm2)接触24小时内培养的balbc 3t3细胞的归一化代谢活性与阳性对照(ctl )的归一化代谢活性来估计直接细胞毒性。ctl (
□
)组和处理组之间没有观察到显著差异。生物复制数=4,总技术复制数=20。
[0068]
●
图12表示在与bsa/nabr膜接触24小时期间培养的balbc 3t3小鼠成纤维细胞的显微镜检查。可以在(a)周围和(b)生物材料上方看到成纤维细胞。
[0069]
●
图13表示在白蛋白膜(bsa/nabr 400、bsa/nabr 664和bsa/nacl
2 700,分别为700,分别为)上测量的balbc 3t3成纤维细胞的归一化代谢活性,这些膜在去除培养基后被新鲜转移到空的未使用的孔中。通过比较处理组和未处理组之间balbc 3t3细胞的归一化代谢活性来估计细胞粘附。(*)在处理组和阳性对照(ctl ,
□
)之间观察到显著差异(p《0.05)。(**)在处理组之间观察到显著差异(p《0.05)。生物复制数=3,总技术复制数=12。
[0070]
●
图14表示在37℃与bsa膜接触48小时期间培养的raw巨噬细胞(m/s=25mg/cm2,从左到右:bsa/nabr 400、bsa/nabr 664、bsa/cacl
2 700、bsa/nabr 400lps、bsa/nabr664lps、bsa/cacl
2 700lps)。测量亚硝酸盐(a)和tnf-α(b)浓度以评估巨噬细胞的活化和炎症反应。未处理组(nt)在没有膜和没有lps的情况下培养。在lps处理的对照组(t lps)和lps激活组(lps)的培养基(50ng/ml)中24小时后添加lps。(*)在处理组和nt组之间观察到显著差异(p《0.05)。(**)在处理组和t(lps)组之间观察到显著差异(p《0.05)。生物复制数=3,总技术复制数=12。
[0071]
●
图15表示在水合(水中)bsa/nabr膜上以0.5hz的固定频率、0.01%至100%的应变范围和在室温下进行的幅度扫描测试。a)储能(g',pa)和损耗(g”,pa)模量表示为幅度(应变,%)的函数。b)损耗模量(g”,pa)表示为幅度(应变,%)的函数。g”模量达到最大值(payne效应)。c)储能模量(g',pa)随时间(s)表示。进行了三个连续的幅度扫描测试。
[0072]
●
图16表示使用sem评估bsa/nabr膜的横截面。
[0073]
●
图17表示热处理(80℃,72小时)bsa/nabr材料(实线)和对照bsa/nabr材料(虚线)的酰胺i带中的ft-ir光谱。
[0074]
●
图18表示在受控真空(200、600和800毫巴)下生产bsa/nabr膜。在大气压下制备对照。a)制备的bsa/nabr膜的视觉方面。b)制备的bsa/nabr膜的相对产率(%,白色)、吸水率(%,斑点)和初始膨胀(%,阴影线)。
[0075]
●
图19表示有机溶剂(乙醇(%,v/v)、dmso(%,v/v)、乙腈(%,v/v)和二氯甲烷(%,v/v))对bsa/nabr膜的相对产率(%,白色)、吸水率(%,斑点)和初始膨胀(%,斑点)的影响的研究,这些有机溶剂被掺入bsa/nabr溶液中,然后在37℃蒸发。使用0%溶剂(溶剂/溶液体积比)制备对照批次。
[0076]
●
图20表示用盐cacl2和nabr的不同组合制备的白蛋白膜的相对产率(%,白色)、吸水率(%,斑点)和初始膨胀(%,斑点)。cacl2/bsa的摩尔比设定为400,nabr/bsa的摩尔比在100至1000之间变化。仅用bsa/cacl2制备对照。
163)在dulbecco改良eagle培养基高葡萄糖(dmem)中培养,该培养基含有稳定的谷氨酰胺和丙酮酸钠(dutscher),辅以10%(v/v)胎牛血清(dutscher))和1%(v/v)的青霉素-链霉素溶液100x(终浓度:分别为0.06mg/ml和0.1mg/ml)(dutscher)。使用胰蛋白酶(0.5g/l)-edta(0.2g/l)(dutscher)在37℃下收获细胞5分钟。噻唑蓝四唑溴化物(mtt)购自sigma-aldrich。celltiterviability assay购自promega。
[0094]
在37℃、5%co2、95%湿度下,将raw 264.7小鼠巨噬细胞(tib-71
tm
)在dulbecco改良eagle培养基高葡萄糖(dmem)中培养,该培养基含有稳定谷氨酰胺(sigma-aldrich),辅以5%(v/v)的热灭活胎牛血清(gibco)、青霉素(100u/ml)(sigma-aldrich)和链霉素(0.1mg/ml)(sigma-aldrich)。使用胰蛋白酶(0.5g/l)-edta(0.2g/l)(sigma-aldrich)在37℃下收获细胞5分钟。来自大肠杆菌(k12)的脂多糖(lps)购自invivogen。用于elisa测试的纯化的抗小鼠tnf-α抗体克隆1f3f3d4和生物素化的抗小鼠tnf-α抗体克隆xt3/xt22购自ebioscience/thermofisher scientific。辣根过氧化物酶抗生物素蛋白(avidin hrp)购自jackson。对氨基苯磺酰胺和乙酸购自sigma。n-(1-萘基)乙二胺二盐酸盐购自acros organics。
[0095]
方法
[0096]
制剂(一般程序)
[0097]
制备bsa(100mg/ml)和nabr 1m(摩尔比nabr/bsa=664)在乙酸钠缓冲液(0.2m)中的ph 6溶液。将该溶液置于模具中(底部覆盖有不粘有机硅盘)并在37℃下蒸发7天。将获得的干燥生物材料洗涤以除去盐,并在室温下在蒸馏水中浸泡48小时。然后收集和表征水不溶性膜(bsa/nabr 664)。
[0098]
初始表征
[0099]
盐/白蛋白的摩尔比(式1)和m/s比(式2)用于标记制剂。使用相对产率(式3)、吸水率(式4)和初始膨胀(式5)比较配制的膜。通过在室温下将材料浸入蒸馏水中评估材料的密度(式6)。wbsa代表用于制剂的白蛋白的初始重量。ai代表在蒸发过程中使用的容器的面积。wd表示在蒸馏水中洗涤48小时并在37℃烘箱中干燥过夜后最终干燥膜的重量。vd是通过将材料在室温下浸入蒸馏水中测量的干燥膜的体积。wh表示在蒸馏水中浸泡24小时并使用滤纸去除多余的表面水后处于平衡状态的水合膜的重量。ah是使用电子数显卡尺(tacklife-dc01,精度
±
0.2mm)测量其直径后计算的水合膜表面的面积。
[0100]
■
[0101]
■
[0102]
■
[0103]
■
[0104]
■
[0105]
[0106]
标准化感应电位(sip)测量
[0107]
通过将浓度为1mg/ml的bsa溶解在mq水中制备滴定溶液。通过将浓度为60mg/ml的nabr溶解在mq水中制备滴定溶液。然后使用nabr溶液通过使用流动电流检测测量感应电势来滴定蛋白质的表面电荷。使用m
ü
tek pcd 02检测器。将10ml bsa溶液转移到检测器罐中。然后,在5分钟的平衡时间后,进行以30μl/min的频率连续添加盐水nabr溶液。当测量的电位达到平台时停止测定。
[0108]
bca测定
[0109]
bsa/nabr 664膜(初始bsa浓度=200mg/ml,m/s=105mg/cm2,n=3)各自用4.5ml超纯水洗涤(2x1.5ml(2x30min)然后1x1.5ml(2h))。然后,对于每种冲洗溶液,使用容量瓶将体积调节至5ml。然后通过使用标准范围(20μg/ml-1000μg/ml)的bca测试在制剂中使用的初始溶液中以及在冲洗溶液中确定白蛋白浓度。该测定在96孔板中进行。将试剂(二辛可宁酸/cuso4)添加到溶液中(200μl试剂添加到25μl蛋白质溶液)。然后,将板在37℃下温育30分钟。使用safas xenius xm荧光分光光度计(safas monaco)在室温下进行560nm处的吸光度读数。计算膜中白蛋白的量后,推导出nabr的量。
[0110]
微量分析
[0111]
使用quanta
tm 250esem(fei company,eindhoven,the netherlands)对样品的随机选择区域进行电子激发x射线显微分析,该仪器在10kv的电子加速电压下运行(出射角=35
°
,采集时间=100s,处理时间=7.68μs)。分析了四种bsa/nabr膜:bsa/nabr 400、bsa/nabr664、bsa/nabr 700和bsa/nabr 1400(m/s=113mg/cm2)。使用样品中溴的原子百分比估计nabr的量。
[0112]
流变行为和压缩测定
[0113]
使用配备有直径为2cm的平面移动装置的malvern kinexus ultra 流变仪进行流变表征和压缩测定。使用了水合bsa/nabr 664膜(m/s=105mg/cm2,厚度=1.7mm)。对于振荡方案,使样品受到1pa的受控剪切应力,并根据在25℃下从100hz到0.01hz的振荡频率变化进行测量。对于压缩测定,在25℃下对样品施加0.5n至40n(0.04mm/s)的力变化。每个样品的弹性模量(e)在应变(ε)-应力(σ)曲线(式7)的弹性域内计算。
[0114]
σ=exε
ꢀꢀꢀꢀ
(7)
[0115]
牵引力测定
[0116]
使用配备100n力传感器的instron electropulstm e3000进行牵引力测定。使用了一批六个水合bsa/nabr 664膜(m/s比为110.9mg/cm2)。用冲头切割样品以形成六个具有标准化尺寸的试样(初始有效长度l0=40mm,有效初始宽度l0=10mm,初始厚度e0=1.74
±
0.079mm)。然后在室温下以0.1mm/s的拉伸速度进行拉伸试验。弹性模量(e)在应变(ε)-应力(σ)曲线(式7)的弹性域内计算。
[0117]
在水溶液中的稳定性
[0118]
将bsa/nabr 664膜(m/s=105mg/cm2)以3个批次放置在25ml以下溶解介质中:蒸馏水、生理盐水(0.9%nacl)、酸性介质(ph 3、颜色指示剂:溴百里酚蓝)、碱性介质(ph 10,颜色指示剂:溴百里酚蓝)、1m nacl盐水溶液、1m nabr盐水溶液、乙醇和胰蛋白酶溶液(在pbs缓冲液中稀释的0.125mg/ml)。然后将含有膜的介质在37℃下伴随摇动(180rpm)温育7天。之后,在表征其质量损失(式8)之前用水洗涤膜。
[0119][0120]
ir分析
[0121]
配备有氘代硫酸胰甘氨酸检测器(rt dlatgs)和kbr分束器的vertex 70ftir光谱仪(德国bruker)用于红外测量。d2o溶液用于避免酰胺i带和水在大约1650cm-1
处的强吸收带之间的光谱重叠。将所有样品都放置在两个caf2窗口之间。将样品的ftir光谱在室温下在4000-800cm-1
之间以2cm-1
标称分辨率记录,每个光谱累积扫描128次,扫描速率为10khz,以d2o光谱为背景。在d2o中制备液体样品(bsa、bsa/nabr 400和bsa/nabr 664的溶液),bsa浓度为100mg/ml。将固体样品(bsa/nabr 400和bsa/nabr 664的膜)用d2o水合。
[0122]
通过使用光谱仪软件opus 7.5(德国bruker)进行光谱分析。对于二级结构分析,对去卷积光谱的酰胺i区域(1700-1600cm-1
)进行曲线拟合。在曲线拟合之前,使用低波数(1600cm-1
)和高波数(1700cm-1
)侧的最小值对酰胺i带的光谱进行基线校正。根据最小二乘迭代曲线拟合程序(levenberg-marquardt)使用高斯线形进行去卷积。子带的数量及其位置由去卷积光谱以及光谱的二阶和四阶导数确定。在最后的拟合中,为了尽可能减少残余rms误差(小于0.005),调整了所有带的高度、宽度和位置,同时每次都不允许更改这些参数中的至少一个。最后,比较原始曲线和拟合曲线的二阶导数,保证曲线拟合的准确性。拟合分量的分数面积用于计算不同二级结构元素(α螺旋、β折叠、β转角和无规卷曲)的百分比。
[0123]
saxs ics
[0124]
对干燥的bsa/nabr 664膜以及三个对照:冻干bsa的干粉和bsa的两种溶液(溶液1:113.69mgbsa 36.31mg h2o,溶液2:119.39mgbsa 79.61mg h2o),进行小角度和广角x射线散射分析(saxs和waxs)。测量是在安装在微焦点旋转阳极发生器(micromaxtm-007hf)上的rigaku衍射仪上进行的,该发生器在40kv和30ma下运行。光束被单色化(波长rigaku衍射仪上进行的,该发生器在40kv和30ma下运行。光束被单色化(波长)并用共焦max-flux opticstm(osmics,inc.)和三针孔准直系统聚焦。散射强度被测量为散射矢量q的大小的函数(式9),其中θ是散射角。使用两种不同的配置来覆盖大的散射矢量范围。
[0125]
使用距离样品位置位于d=0.81m的2d多线检测器研究低q范围,使用更靠近样品插入的富士成像板测量更高的q值根据各向同性散射的常用程序处理散射图案:对强度进行径向积分并针对电子背景、检测器效率、样品透射率和样品厚度进行校正。对于bsa溶液,还测量并减去来自纯溶剂和容器的散射。使用校准的lupolen标准将强度转换为绝对比例。使用山嵛酸银粉末的衍射峰校准散射矢量。
[0126][0127]
扫描电子显微镜(sem)
[0128]
使用quanta
tm 250esem(fei company,eidhoven,the netherlands)进行扫描电子显微镜评估,在电子加速电压10kv下操作。bsa/nabr 664膜(m/s比=105mg/cm2)在37℃下干燥48小时。然后使用hummer jr溅射装置(technics,union city,ca,usa)将样品涂上金钯合金。检查表面和横截面。
[0129]
提取物细胞毒性试验
[0130]
对于间接细胞毒性评估,遵循iso标准(iso 10993-5(2009))。该测试中使用的膜是bsa/nabr 400、bsa/nabr 664和bsa/cacl
2 700。膜以25mg/cm2的m/s比率配制。将膜用70%乙醇洗涤,然后用无菌pbs 1x洗涤,并在紫外光下灭菌15分钟。然后,将它们储存在无菌pbs 1x中,直到进一步使用。将每个膜转移到12孔板中,并在37℃搅拌下在1.5ml培养基(dmem fbs(10%) ps(1%))中提取72小时。然后制备含有12.5%、25%、50%和100%(v/v)提取物的稀释液。将balbc 3t3小鼠成纤维细胞(克隆a31ccl-163)在96孔板中以每孔8000个细胞(培养基:dmem fbs(10%) ps(1%)在37℃下培养24h。第二天,每孔中的培养基用100μl的稀释提取物替换。仅用培养基和用含有20%dmso的培养基分别制备阳性和阴性对照。然后将板在37℃下温育24小时。温育后,将每孔中的培养基更换为100μl用新鲜培养基(1mg/ml)稀释的mtt溶液,并将板在37℃下温育2h。然后将甲臜晶体溶解在80μl dmso中,在室温下平衡15分钟后,使用safas装置测量560nm处的吸光度。阳性对照的代谢活性用于确定每组中活细胞的百分比。
[0131]
直接细胞毒性试验
[0132]
该测试中使用的膜是bsa/nabr 400、bsa/nabr 664和bsa/cacl
2 700。在不粘硅树脂模具中以25mg/cm2的m/s比配制膜。使用圆形冲头切割水合膜以获得小圆片(直径=5mm,厚度=0.7mm)。将圆片用70%乙醇洗涤,然后用无菌pbs 1x洗涤,并在紫外光下灭菌15分钟。然后,将它们储存在无菌pbs 1x中,直到进一步使用。对于直接细胞毒性评估,将灭菌的圆片转移到黑壁96孔板中。然后将balbc 3t3小鼠成纤维细胞(克隆a31ccl-163)以每孔8000个细胞(培养基:dmem fbs(10%) ps(1%))直接添加到生物材料圆片上。仅用培养基和用含有20%dmso的培养基分别加入阳性和阴性对照。然后将板在37℃温育24小时。温育后,将板在室温平衡30分钟。将培养基去除。在每个孔中,加入50μl新培养基,然后加入50μl试剂。然后使用safas设备和以下方案测量生物发光:将板搅拌2分钟,然后在测量生物发光之前保持平衡10分钟。阳性对照的代谢活性用于确定每组中活细胞的百分比。
[0133]
巨噬细胞活化试验
[0134]
该测试中使用的膜是bsa/nabr 400、bsa/nabr 664和bsa/cacl
2 700。在不粘硅树脂模具中以25mg/cm2的m/s比配制膜。使用圆形冲头切割水合膜以获得小圆片(直径=5mm,厚度=0.7mm)。将圆片用70%乙醇洗涤,然后用无菌pbs 1x洗涤,并在紫外光下灭菌15分钟。然后,将它们储存在无菌pbs 1x中,直到进一步使用。对于巨噬细胞活化测定,将灭菌的圆片转移到96孔板中。然后将raw 264.7巨噬细胞以每孔100000个细胞(培养基:dmem fbs(5%) ps(1%))直接添加到生物材料圆片上。在37℃下温育24小时后,将lps添加到lps处理组中,以在每个孔中获得50ng/ml的最终浓度。然后将板在37℃下再温育24小时。仅用培养基和用含有50ng/ml lps的培养基分别制备阴性和阳性对照。然后通过显微镜评估细胞的形状并且如下评估no和tnf-α的产生。
[0135]
no产生的评估
[0136]
通过griess测试(n=3)评估细胞上清液中亚硝酸盐的浓度。将60μl的griess试剂(30%乙酸中的58.1mm对氨基苯磺酰胺和60%乙酸中的3.9mm n-(1-萘基)乙二胺二盐酸盐的v/v混合物)添加到40μl上清液中,并测定在543nm处的吸光度并与亚硝酸钠标准曲线进
行比较。
[0137]
评估tnf-α的产生
[0138]
使用市售试剂并按照制造商说明通过elisa(n=3)评估细胞上清液中的tnf-α浓度。将捕获抗体在0.05m ph 9.6碳酸盐/碳酸氢盐缓冲液中稀释至1μg/ml,并在4℃下包被1晚,然后用pbs 0.05%tween 20 1%bsa封闭(1h,37℃)。然后用培养基稀释样品并与捕获抗体一起温育(2h,37℃),然后加入在pbs 0.05%tween 20 1%bsa中稀释至0.5μg/ml的检测抗体(1h,37℃)。然后引入抗生物素蛋白hrp(45分钟,37℃)并通过添加1.25mm四甲基联苯胺和13.05mm h2o2在0.1m ph 5柠檬酸盐缓冲液中的溶液进行揭示。最后通过添加1m hcl停止揭示,并在450nm处测量吸光度。
[0139]
统计分析
[0140]
使用r(3.6.1版,r foundation for statistical computing,vienna,austria)分析数据。分布的正态性由shapiro-wilk检验确定。用f检验确定方差的相等性。当数据呈正态分布且样本方差相等时,使用t检验(2尾)比较两个均值。当这两个条件不适用时,改为执行mann-whitney检验。在p《0.05时值被认为具有统计学意义。
[0141]
结果和讨论
[0142]
制剂参数筛选
[0143]
彻底研究了白蛋白在水溶液中的溶解度及其热稳定性和ph稳定性区域。先前的研究表明,白蛋白在3到9的ph值范围内是稳定的。还表明,变性温度取决于ph值,并在低ph值时降低(ph 7.4时为62℃,ph 3.5时为46.8℃)。在这项工作中,蒸发温度设置为37℃,ph设置为6,以尽可能地保留白蛋白的天然结构。然后,对操作条件进行了彻底筛选,以确定那些允许形成有趣的生物材料的条件。将在受控ph(ph=6)下配制的白蛋白溶液(单独的蛋白质或加盐)在37℃的烘箱中蒸发,直到残留物完全干燥(见图1)。之后,洗涤残留物以去除多余的盐和蛋白质,然后在蒸馏水中浸泡48小时以评估它们的水溶性。仅选择产生水不溶性材料的制剂。对于生物材料领域的潜在应用,表现出良好可操作性的水不溶性膜是最有前途的。
[0144]
对于以105mg/cm2的m/s配制的材料,在68至69小时后获得固体材料(见图2)。69小时后,随着残留水分的蒸发,材料表面会形成一层薄薄的白色过量盐分。洗涤后,盐层迅速消除,得到半透明膜(见图2)。发现7天的蒸发持续时间对于m/s为105mg/cm2的膜是最佳的,以允许材料的形成和压实。
[0145]
第一个参数:盐
[0146]
首先,验证了无盐制备的白蛋白残留物的溶解度,证实了盐在白蛋白膜制剂中的重要性。然后,对三种不同浓度(0.5m、1m和2m)的许多盐进行了彻底筛选。如前所述,干燥残留物在蒸馏水中48小时后的水溶性用于鉴定允许膜形成的盐。
[0147]
根据hofmeister系列,不同的离子对蛋白质的稳定性和溶解度有不同的影响。易溶效应与离子的大小、电荷密度和极化率有关。当使用含有高浓度盐和蛋白质的溶液时,应考虑溶剂分子、盐离子和蛋白质之间相互作用的影响。在该实验中,测试了12种具有各种阴离子和阳离子对的盐:kcl、kbr、ki、nacl、nabr、nai、cacl2、mgcl2、kc2h3o2、nh4hco2、k2co3和k2hpo4。该实验的结果表明,白蛋白膜的形成取决于盐的类型及其浓度。在kcl、nacl或kbr的存在下,白蛋白分子不会组织成膜,干燥的残留物是完全溶于水的结晶盐和干燥蛋白质的
混合物。此外,用含有二价阴离子的盐(k2co3和k2hpo4)制备的初始溶液中白蛋白的过早聚集阻止了膜的形成。用12种盐中的7种获得水不溶性膜:nabr、ki、nai、cacl2、mgcl2、kc2h3o2和nh4hco2。使用nabr,获得了所有三种测试浓度的膜。虽然,对于其他6种盐,白蛋白膜仅在一定浓度下获得。这些膜的物理方面和特性(吸水性、初始膨胀、可操作性)根据所用盐的类型及其浓度而有很大差异。
[0148]
bsa/nabr 664膜(初始盐浓度=1m)表现出良好的机械强度和可操作性。这些膜(m/s=105mg/cm2)的相对产率为87.6%
±
4.1%,其吸水率和初始膨胀率估计分别为123.1%
±
6.8%和121.6%
±
4.7%(n=8),其密度为1.29
±
0.02g/cm3。选择该制剂作为确定基于白蛋白的膜形成的参数和表征这些材料的参照。
[0149]
第二个参数:盐/白蛋白摩尔比
[0150]
先前已表明盐浓度对膜形成有影响。然而,尚不清楚盐的浓度是否应被视为独立参数,还是应与给定溶液中的白蛋白浓度配对。bsa和nabr的初始浓度对膜形成的影响是通过比较在两次测定中获得的膜评估的:第一次测定涉及恒定浓度的nabr和可变浓度的bsa,因此盐/白蛋白的摩尔比可变,而第二次测定需要同时修改两种浓度而不改变盐/白蛋白的摩尔比。在第一次测定中,用1m nabr制备100mg/ml、200mg/ml、300mg/ml和400mg/ml bsa的溶液。nabr/bsa摩尔比分别为664、332、221和166。降低摩尔比导致制剂产率显著降低(分别为86.4%、77.2%、62.7%和0%),并且获得的膜在视觉方面和吸水性方面差异很大。此外,白蛋白浓度为400mg/ml(盐/白蛋白摩尔比=166)的溶液蒸发后得到的残余物完全溶于水。在第二次测定中,制备的溶液分别含有以下浓度的bsa和nabr:100mg/ml与1m、200mg/ml与2m、300mg/ml与3m和400mg/ml与4m。在所有溶液中,nabr/bsa的摩尔比为664。与第一次测定不同,第二次测定的所有溶液都导致膜形成。获得的膜具有相同的视觉方面并表现出相似的特性。因此,如果在初始溶液中不将盐浓度与白蛋白浓度配对,就无法评估盐浓度对膜形成的影响。由于膜的形成取决于盐和白蛋白浓度的配对效应,因此盐/白蛋白的摩尔比被证明是评估白蛋白膜形成的更相关和可靠的参数。
[0151]
下一步是确定可以形成膜的nabr/bsa摩尔比范围。bsa溶液是用设定浓度的白蛋白(100mg/m)和50至2000的nabr/bsa摩尔比制备的。如前所述,蒸发这些溶液并将所得材料浸泡在水中48小时。在摩尔比100和3000的范围内,特别是在200到2000的范围内,获得了完全形成的膜。对于该范围的最低和最高摩尔比,所获得的膜不太坚固并且在处理过程中更容易破裂和降解,但是它们是可以接受的。
[0152]
白蛋白表面电荷的影响
[0153]
由于初始溶液的离子含量对白蛋白膜制剂的明确影响,应评估白蛋白的表面电荷,以更好地了解导致膜形成的离子现象。白蛋白的表面电荷取决于初始溶液的ph值及其离子强度。
[0154]
为了评估ph值对膜形成的影响,在4、5、6、7和8的ph值下制备了nabr/bsa摩尔比为664的白蛋白和nabr溶液。bsa具有4.7的等电点和5.2的等离子点,在ph 6、7和8时具有净负电荷,在ph 4时具有净正电荷,并且在ph 5附近是两性离子。在所有测试的ph值下获得白蛋白膜。这些膜具有相似的视觉外观和制剂产量,但它们的吸水率和初始膨胀显著不同。在最高ph值下配制的膜具有更高的吸水率和初始膨胀率。因此,ph似乎对膜的形成有适度的影响。此外,在ph值为6时对白蛋白表面电荷的研究表明,通过添加盐,由于蛋白质与盐的阳离
子之间的相互作用,蛋白质的整体表面电荷会增加。事实上,通过在达到平台之前增加盐浓度,测量的感应电势会大大增加(见图3)。白蛋白表面电荷的增加可导致分子之间的静电排斥减少并促进它们聚集形成白蛋白膜。
[0155]
残留盐含量
[0156]
在进行生物学评估时,应充分表征配制材料中的最终成分和残留盐含量。因此,为了确定bsa/nabr 664膜的最终成分并量化残留nabr,使用了两种互补的方法。首先,使用bca测定法,对用于清洗bsa/nabr 664膜的冲洗水中的白蛋白进行定量。冲洗溶液蒸发后,称重干燥残留物,计算通过洗涤过程消除的溴化钠量,并与最初用于配制膜的溴化钠量进行比较。经洗涤的bsa/nabr 664膜中的残留nabr含量估计小于1%(重量%)。为了验证这些结果并特别针对溴含量,通过微量分析直接分析了bsa/nabr 664膜的最终组成。直接对干燥的膜进行微量分析,表明在分析区域(分析区域=1180μm2,样品厚度=1mm)中检测不到测试膜中的溴含量。此外,x射线衍射对bsa/nabr膜的分析(见图8a和图8b)在膜中没有检测到任何痕量的结晶nabr。总之,最初添加到溶液中用于配制基于白蛋白的生物材料的溴化钠在洗涤过程中被去除。
[0157]
机械性能
[0158]
研究了所选的基于白蛋白的生物材料在用水饱和后的粘弹性行为。发现守恒模量(g')高于损耗模量(g”)(见图4)。此外,没有观察到溶胶-凝胶转变。因此,在测试的频率范围内,膜表现出类似固体的行为。
[0159]
在压缩测定过程中,发现用摩尔比为664的bsa和nabr配制的膜具有0.7mpa的弹性模量。此外,对以相同方式配制的一批白蛋白膜(n=6)进行了拉伸试验。牵引测定后计算的膜的弹性模量为0.87
±
0.12mpa。因此,两种测定都给出了相似的结果。导致生物材料断裂的最大应力估计为0.19
±
0.03mpa。
[0160]
下面的表1显示了牵引和压缩结果之间的比较。使用的膜在选定的条件下(在37℃和ph=6下蒸发)用bsa(牛血清白蛋白)和nabr配制,nabr/bsa摩尔比为664。测试是在水合生物材料上进行的。
[0161]
表1:
[0162] 牵引力测定压缩测定初始厚度(mm)1.74
±
0.081.7断裂应变(%)26.2
±
4.7无断裂最大应力(mpa)0.19
±
0.03》0.13最大力(n)3.2
±
0.5》40弹性模量(mpa)0.86
±
0.130.7
[0163]
在水溶液中的稳定性
[0164]
重要的是用于与生物流体接触的生物材料在水性介质中具有良好的稳定性。对于用于组织再生的植入物或支架的制剂等应用,生物材料应不溶于水或具有非常缓慢的降解过程。因此,测试了膜bsa/nabr 664在水溶液中的稳定性。还在胰蛋白酶溶液中测试了膜的生物降解性(见图5)。
[0165]
与胰蛋白酶溶液接触温育的膜完全降解并溶解在缓冲液中。对于在没有蛋白酶的情况下在37℃下温育7天的膜,每批膜在每种溶出介质中的损失小于其初始质量的10%,但
在碱性溶液中温育的膜除外,其损失了其质量的17%。此外,配制的基于白蛋白的膜不溶于水性介质,在37℃下在这些介质中温育7天以上仍保持完整。事实上,这些膜可以在蒸馏水中储存长达一个月,而不会显示任何降解。此外,这些生物材料显示出对酸性和碱性ph值的抵抗力。在水和乙醇中观察到的质量损失主要可以解释为在搅拌过程中由于管侧面摩擦引起的膜腐蚀。因此,这些基于白蛋白的膜在水溶液中非常稳定并且完全可生物降解。
[0166]
还测试了bsa/nabr 664膜在尿素(2m、4m和8m)和2-巯基乙醇(0.1m)溶液中的稳定性。尿素和2-巯基乙醇是众所周知的变性剂,它们可以分别通过破坏氢键和二硫键来诱导蛋白质展开。尽管测试的基于白蛋白的膜没有破裂或溶解,但水合膜的直径(ah)显著增加,表明它们的初始膨胀(e%)增加,因为它们的复数模量(g*)降低,表明它们的弹性模量(e)降低(见图6)。此外,氢键和二硫键的断裂改变了膜的结构,但不允许它们溶解。
[0167]
白蛋白构象的评价
[0168]
ir分析
[0169]
傅里叶变换红外光谱(ftir)是一种成熟的方法,用于评估蛋白质的二级结构。蛋白质的红外光谱的特征在于吸收光谱中的一组吸收区域,分别称为酰胺区域和ch区域。关于二级结构的信息可以从主要来自酰胺i区域(1700-1600cm-1
)和酰胺ii区域(1600-1500cm-1
)的光谱中获得。酰胺i区域主要反映肽基团的c=o伸缩振动,它提供了蛋白质二级结构的信息。此外,该技术允许探索高浓度溶液和固体材料中所含蛋白质的二级结构,并可用于水合或干燥材料。ftir用于评估bsa/nabr膜中白蛋白的结构。在本实验中,为防止h2o带干扰位于相同吸收范围内的酰胺i带,使用d2o水合易得的bsa/nabr 664膜并制备两种对照溶液:bsa(100mg/ml)的溶液以及bsa和nabr的溶液(bsa浓度=100mg/ml,bsa/nabr比率=1:664),其与用于配制bsa/nabr 664膜的溶液相当。
[0170]
对酰胺i带的去卷积光谱的分析揭示了多个子带的存在,这些子带是使用科学文献中已有的数据确定的。在测试样品的酰胺i带中发现了六个子带:α-螺旋(1655cm-1
)、β-折叠(1612、1629和1678cm-1
)、β-转角(1669cm-1
)和无规卷曲(1643cm-1
)子带(见图7)。然后计算这些二级结构的百分比(残余rms误差《0.005)。对于bsa和bsa/nabr 664的溶液中存在的白蛋白,获得了非常相似的β-折叠、β-转角、α-螺旋和无规卷曲的百分比。因此,在既定的实验条件下,nabr不会改变白蛋白的二级结构。然而,在bsa/nabr 664膜中,似乎β-折叠和β-转角增加,而α-螺旋和无规卷曲减少(表2)。蛋白质的展开的特征在于无组织二级结构的无规卷曲的增加。在膜的制剂中,白蛋白似乎对这些无组织结构中的一些有利于β-组织结构。
[0171]
下表2显示了在bsa溶液(100mg/ml,在d2o中)、bsa/nabr 664溶液(bsa浓度=100mg/ml,在d2o中)和bsa/nabr 664膜(残余rms误差《0.005)中酰胺i带中确定的每个二级结构的百分比的分析。bsa溶液(bsa 100mg/ml 80℃d2o)在80℃下温育过夜,并用作变性蛋白质的参照。
[0172]
表2:
[0173] α螺旋β折叠β转角无规卷曲分子间β折叠bsa/nabr 664溶液d2o31.7%28.5%6.0%33.8%3.8%bsa/nabr 664膜d2o21.8%46.8%16.5%14.8%23.6%bsa 100mg/ml d2o30.6%28.2%6.9%34.3%2.9%bsa 100mg/ml 80℃d2o8.8%48.8%5.7%36.7%2.6%
[0174]
晶体学saxs ics
[0175]
小角度和广角x射线散射测量(saxs、waxs)探测样品内的原子间距离。对于溶液中的蛋白质,它们揭示了大分子的层次结构层级。因此,不同的散射矢量域与以下有关:分子的特征组织,例如四级和三级结构(分子的形状和大小,),域间相关性),域间相关性域内组织和二级结构waxs对结构紊乱或波动非常敏感,能够证明大分子组织的微小变化。然而,数据的解释非常复杂,通常需要与基于晶体学结果的理论计算进行比较。在不存在分子间相关性的稀释状态下,散射测量仅反映分子内特性(形状因子)。当浓度增加或大块膜特性增加时,情况不再如此,分子间相关性对散射强度有很大贡献。
[0176]
bsa/nabr 664膜的结构组织通过saxs和waxs探测。测量洗涤和干燥的膜(e,图2中.s8b)并与天然bsa(a,图2中.s8b)进行比较。不同浓度下制备的两种bsa溶液(b和c,图2中.s8b)(溶液1 24.2重量%h2o,溶液2 40重量%h2o)也被认为为解释散射模式提供了参考。实验强度显示在图2中.s8a(溶液2)中,并与使用crysol计算的理论曲线进行比较,并基于单体bsa的晶体学描述(pdb原子坐标3v03)。在0.1和之间,实验曲线和理论曲线都很好地对应。小的偏差当然反映了溶液中蛋白质的动态波动和无序。在以下,在附近观察到峰。它与计算中未考虑的静电排斥导致的bsa之间的空间相关性有关。通过增加蛋白质浓度(溶液1),由于分子质心之间的距离减小,该峰向更高的q值移动。在高q值下,在附近观察到较大的最大值。在这个区域,模型不太准确,但这超出了程序可接受的q范围因此,crysol能够解释溶液中bsa数据的主要特征。由于蛋白质主要包含处于结晶有序状态的α-螺旋(如计算中所用),因此在附近观察到的预测的肩峰可能与α域之间的相关性有关,而在0.4和之间的三个贡献(9
→→
)与域的内部结构有关,更具体地说与α-螺旋堆积有关。对于所有蛋白质,发现了理论上没有正确描述的附近的最大宽度并且该最大宽度对内部组织的主要特征(α-螺旋或β-折叠)不太敏感。
[0177]
冻干粉末bsa的散射模式如图2中.s8b(a)所示。这种无定形样品用作非溶剂化天然bsa的参照。与简单的bsa溶液相比,强度被高度修改。数据解释更加复杂,因为分子内和分子间的相关性现在归因于散射强度。无序、分子间相关性或特定重排可以解释溶液和固态之间散射曲线的潜在变化。不再可能确定蛋白质的形状和大小,只能测量与内部结构或分子间相关性相关的较短长度尺度。首先在非常低的q观察到大的回升。这种行为与任何特定组织无关,只是样品粉末性质的结果(porod散射)。在没有这种贡献的情况下,强度将趋于恒定的低值(q
→
0)。在范围0.1
→
内,在附近观察到小的最大值。该峰可能与溶液中“球形”bsa分子观察到的相关峰的高浓度限制有关。然而,这似乎不太可能,因为接触的蛋白质之间的平均距离将非常接近它们的回转半径该最大值更可能与蛋白质结构域之间的相关性(包括分子内和分子间贡献)相关,并且相当于之前在bsa溶液中观察到的约的肩峰(纯分子内贡献)。因此,α-结构域仍以固态存在。在0.3和之间观察到其他变化。bsa溶液中的结构化块现在减
3t3小鼠成纤维细胞来评估膜可浸出成分的细胞毒性。对于这种细胞系,可以通过测量代谢活性来估计细胞活力。然后将用每种提取物培养的细胞的归一化代谢活性与未处理细胞(阳性对照)的代谢活性进行比较。对于这个实验,提取物被稀释以显示任何剂量依赖性效应。所得数据的统计分析得出结论,未处理组的代谢活性与用不同稀释度的bsa/nabr膜提取物处理的组之间没有显著差异(见图10)。此外,如前所示,白蛋白的非细胞毒性和最终膜中不存在nabr与观察到的bsa/nabr膜的可浸出组分的非细胞毒性相匹配。然后,通过直接用这些膜温育balbc 3t3细胞评估直接细胞毒性。在该实验中使用了相对薄的膜(平均厚度≈0.7mm),以便通过显微镜观察细胞与材料之间的相互作用(见图11)。数据的统计分析表明,未经处理的细胞和用测试膜培养的细胞之间的代谢活性没有显著差异(见图10)。此外,显微镜检查显示成纤维细胞在膜周围和膜上方扩散(见图12)。为了量化膜上的细胞粘附,balbc 3t3细胞与膜一起温育24小时。然后,除去培养基,将膜转移到空孔中并测量每个膜上细胞的代谢活性。在bsa/nabr 664、bsa/nabr 400和bsa/cacl
2 700组之间观察到显著差异(p《0.05)。大约45%(44.55%
±
10.67%)的成纤维细胞附着在bsa/nabr 664膜上。bsa/nabr 400膜的估计百分比较低(27.36%
±
11.22%),而bsa/cacl
2 700膜的估计百分比较高(77.62%
±
20.26%)(见图13)。因此,尽管在洗涤过程中完全消除了盐分,但初始制剂溶液似乎对细胞与膜的相互作用具有显著影响。总之,配制的基于白蛋白的生物材料是无细胞毒性的,有利于细胞粘附和定植。
[0187]
巨噬细胞活化
[0188]
通过测量亚硝酸盐和tnf-α浓度来评估白蛋白膜对巨噬细胞活化的影响。no和tnf-α由活化的巨噬细胞产生,以启动和维持炎症反应。用测试的膜培养原始巨噬细胞24小时。之后,将lps直接引入lps激活组的孔中以激活巨噬细胞,并将细胞再培养24小时。与未处理组(nt)相比,在测试膜存在的情况下,亚硝酸盐的产生略有增加(p《0.05)。lps活化诱导培养基中亚硝酸盐浓度的显著增加。然而,与lps处理的对照(t lps)相比,当巨噬细胞与白蛋白膜一起培养时,亚硝酸盐的产生似乎显著降低(p《0.05)。灭活组的tnf-α产生遵循类似的趋势。虽然,在bsa/nabr组和t lps组中tnf-α的产生没有显著差异,这与bsa/cacl
2 700组的tnf-α产生显著降低不同(p《0.05)。因此,测试的基于白蛋白的膜不能通过激活巨噬细胞有效地诱导炎症反应。
[0189]
结论
[0190]
在这些测试中,使用以下盐开发了几种有趣的生物材料模型:nabr、nai、ki、cacl2、mgcl2、乙酸钾和甲酸铵。这些膜的特性可以通过修改制剂参数来调节。确定了两个重要参数:盐的存在和盐/白蛋白的摩尔比。此外,还可以获得具有三元系统盐1/盐2/白蛋白或盐/白蛋白/聚合物的膜,它们已成功配制。这些系统可以调节膜的特性(机械和内在特性)并获得具有新特性的功能化生物材料。在盐存在下通过蒸发配制的基于白蛋白的生物材料形成了一种多功能模型,其特性可以调节以适应靶向治疗应用的要求。
[0191]
实施例2:物理化学研究和评估该技术的通用性,活性物质的加载,以及生物相容性和体内生物降解性的初步评估
[0192]
2.1物理化学调查和评估技术的通用性
[0193]
对于以下实验,通过在37℃蒸发在乙酸钠缓冲液(ph=6)中制备的bsa(初始浓度=100mg/ml)和nabr(初始浓度=62mg/ml)的溶液,直到形成干燥的材料,制备bsa/nabr材
料。对于bsa/cacl2材料,通过在37℃蒸发在乙酸钠缓冲液(ph=6)中制备的bsa(初始浓度=100mg/ml)和cacl2(初始浓度=155mg/ml)的溶液,直到形成干燥的材料,制备它们。
[0194]
在有机溶剂中稳定性.大多数活性物质是亲脂性分子,有机溶剂对于它们在生物材料中的溶解和加载非常有用。此外,在容易形成的材料中加载药物(后加载)需要材料在用于溶解药物的溶剂中的稳定性。albupad材料(即根据本发明的生物材料)在有机溶剂中的稳定性在以下溶剂中进行评估:乙醇、dmso、乙腈和二氯甲烷。将bsa/nabr和bsa/cacl2膜在室温下在每种溶剂中放置72小时。通过比较它们的质量损失来评估它们的稳定性。在所有测试的溶剂中温育72小时后,膜没有出现质量损失。此外,它们的物理方面没有显示出任何明显的降解迹象,并且它们的水合特性也得以保留。因此,albupad材料在乙醇、dmso、乙腈和二氯甲烷中是稳定的。
[0195]
bsa/nabr膜的流变学评估.在这项研究中,对水合(在水中)bsa/nabr膜(n=4)进行了幅度扫描测试,以研究它们的粘弹性行为。g'代表弹性或可恢复分量,g”是粘性分量。在每次测试期间,频率设置为0.5hz,而应变从0.01%增加到100%。超过10%的应变观察到强烈的滑动效应,在任何测试样品中都没有任何明显的损坏,导致数据无法使用(省略)。在1%应变下的线性粘弹性区域(lve)中,g'(59.6
±
5.1kpa)高于g”(7.7
±
0.8kpa)(图15中a)。因此,该材料在该范围内表现为固体弹性材料。在1%应变后,随着g”增加,确定了payne效应,并在1.79
±
0.07%应变时达到最大值(图15中b)。这种效应是由两相组成的材料的特征,其中较硬的颗粒悬浮在基质中,在某些变形时会导致更大的能量耗散。这种效应主要在载有炭黑颗粒的橡胶弹性体中进行了描述,并归因于材料微观结构变形引起的变化。因此,bsa/nabr材料由含有较硬颗粒的基质组成。然而,已确定盐在洗涤过程中被消除。因此,这些颗粒很可能是围绕在较软的白蛋白基质中的白蛋白聚集体/颗粒。该结果通过对bsa/nabr材料截面的meb分析得到证实,揭示了形成这些材料的颗粒结构(图16)。当材料经过多次连续幅度扫描测试(图15中c)时,g'恢复其初始值,证明材料的内聚力在实验过程中没有改变。对其他几种制剂(例如bsa/cacl2膜)进行了类似的观察,表明这些流变学发现是albupad材料的特性特征。
[0196]
bsa/nabr膜的接触角.通过在bsa/nabr膜的干燥样品表面上沉积一微滴milliq水(5μl)来进行接触角测量(沉积速率=2μl/s)。图像记录100秒(1.4fps)。在这些测试期间,测量了在90
°
和100
°
之间变化的接触角。膜的水合动力学和液滴沉入材料的动力学很慢(》30分钟),因此在这些测量过程中无法观察到。因此,干燥的bsa/nabr膜的表面由于缓慢的水合动力学和高表面粗糙度而具有适度的亲水性(在之前的工作中通过sem分析观察到)。
[0197]
bsa/nabr膜的加速老化.将干燥的bsa/nabr膜置于80℃的烘箱中3天。然后使用ft-ir分析热处理膜的酰胺i光谱,并与对照膜进行比较。热处理膜的酰胺i带与未处理的对照相似(图17)。因此,在测试条件下加热不会改变膜中白蛋白的二级结构。
[0198]
在受控真空下生产.在真空下通过蒸发生产albupad材料可以提供减少蒸发所需的时间的有用工具。因此,在受控压力下的制剂的可行性是使用真空烘箱进行的。将bsa/nabr溶液(bsa的初始浓度=100mg/ml,nabr的初始浓度=1m)在真空烘箱中于37℃下蒸发24小时,并测试以下压力值:800、600和200毫巴。然后将干燥的材料洗涤并在水中浸泡48小时以评估其稳定性。此外,物理外观、相对产率、吸水率和初始膨胀值用于表征在受控真空下形成的材料,并将它们与在大气压下制备的对照批次进行比较。在所有测试的压力值下,
在受控真空下蒸发bsa/nabr溶液后,获得了可处理和不溶于水的材料。所形成的膜具有与对照批次相似的物理特性和可操作性。然而,在膜形成过程中从溶液中消除可溶性气体导致膜结构中的气泡被截留,导致在200和600毫巴下形成大孔(图18中a)。因此,需要优化配方以控制或防止这些孔的形成,例如通过在蒸发之前添加溶液脱气步骤。此外,真空下制剂的相对产率(60%至70%)相对低于对照批次(》90%)(图18中b)。接下来,评估了在600毫巴下制备的膜的机械和流变性能。使用平行板测量系统中的流变仪对水合bsa/nabr 600毫巴水合(在水中)膜进行压缩测定。膜的弹性模量为0.76mpa,与对照批次的弹性模量和先前测量的bsa/nabr材料的弹性模量相似。对水合(水中)bsa/nabr膜(n=4)进行幅度扫描测试,以研究它们的流变特性。在每次测试期间,频率设置为0.5hz,而应变从0.01%增加到100%。这些实验表明,在600毫巴的受控真空下配制的bsa/nabr膜与在大气压下配制的bsa/nabr膜具有相似的流变特性。因此,受控真空下的制剂可以在不改变其特性的情况下形成albupad材料。
[0199]
溶剂调查.使用有机溶剂(如乙醇、dmso或乙腈)可以提供一种有趣的工具,以将水溶性差的活性物质掺入白蛋白和盐的溶液中,从而提供一种将这些分子预加载到albupad材料中的方法。在蒸发步骤(对于挥发性溶剂,如乙醇)或洗涤步骤(对于与水混溶的溶剂,如dmso),容易除去有机溶剂。因此测试了在混合有机溶剂/白蛋白溶液中配制albupad材料的可行性研究。在本次调查中,选择了四种溶剂:乙醇、dmso、乙腈和二氯甲烷。根据以下溶剂/溶液体积比将溶剂直接添加到bsa/nabr溶液中:2.5、5、10、15、20、25和30%v/v。然后将溶剂/溶液混合物在37℃蒸发7天。随后,将干燥的材料在蒸馏水中洗涤48小时。将配制材料的相对产率、吸水率和初始膨胀与没有有机溶剂配制的对照批次(体积比=0%)进行比较。在所有测试比例的四种溶剂存在下形成稳定和水不溶性膜。这些材料是可处理的,并且与对照批次具有相似的物理特性。对于在乙醇存在下配制的材料,以2.5%至15%的体积比制备的膜具有与对照批次相似的相对产率(85
–
88%)、吸水率(238
–
250%)和初始膨胀(137
–
145%)值(图19)。与对照批次相比,以20%至30%的体积比制备的膜显示出相对产率下降(80
–
82%)吸水率(304
–
346%)和初始膨胀值(147
–
154%)增加(图19)。dmso的存在对制剂的相对产率或材料的初始膨胀值没有影响。以2.5%至10%的体积比制备的膜的吸水率值与对照批次相当;同时,体积比在15%到30%范围内制备的膜的吸水率值更高(194
–
345%)(图19)。至于测试的非水混溶性溶剂(乙腈、二氯甲烷),所有制备的膜具有相似的相对产率、吸水率和初始膨胀值(图19)。总之,高达30%(v/v)的有机溶剂的存在不会阻止膜的形成,并且可以制备可处理的材料。与高达15%乙醇、10%dmso和30%乙腈或二氯甲烷的对照批次相比,未观察到这些材料的性能发生任何变化。在bsa/cacl2膜以及bsa/nabr和bsa/cacl2海绵的情况下获得了类似的结果。
[0200]
盐的组合.盐是使用albupad技术配制白蛋白材料的关键参数,盐的类型及其浓度是调节材料性能的相关工具。此外,盐组合可以为更好地调整材料特性提供额外的工具,使albupad材料平台能够根据潜在应用的要求更加灵活和适应性强。因此,研究了albupad技术对使用盐组合配制白蛋白材料的适用性。对于这些实验,测试了两种不同盐的各种组合:一级盐(s1)和二级盐(s2)。设定s1的盐/白蛋白摩尔比,而s2的盐/白蛋白摩尔比从100变化到1000。测试了以下盐的组合:nabr 400/cacl2、cacl
2 400/nabr、nabr 400/mgcl2、nabr50/nacl、nabr 400/nacl、cacl
2 400/mgcl2和nacl 400/kcl。在所有测试的s2摩尔比下,使用组
合nabr 400/cacl2、cacl
2 400/nabr、nabr 400/mgcl2、nabr 400/nacl、cacl
2 400/mgcl2获得了可处理的膜。在测试的盐中,盐nabr、cacl2和mgcl2允许形成可处理的和稳定的白蛋白材料。事实上,如果盐的组合包括至少一种允许膜形成的盐并且如果该盐的盐/白蛋白的摩尔比足够(》100),则获得白蛋白膜。使用相对产率、吸水率和初始膨胀来表征形成的材料,以评估二级盐对材料性能的影响。图20表示使用cacl
2 400/nabr组合配制的膜。测试了100
–
1000范围内的nabr摩尔比,并制备了仅用cacl2和bsa以400的盐/白蛋白摩尔比制备的对照。在制剂中添加溴化钠不会改变制剂的相对产率。然而,它使吸水量显著增加。其他组合也注意到膜性能的改变。此外,还测试了三种盐的组合的制剂的可行性。这些实验表明,使用nabr/cacl2/mgcl2组合(测试的摩尔比:100/100/100、200/200/200、300/300/300)可以生产稳定且易于处理的膜。总之,使用多种盐不会阻止膜的形成,并为albupad材料性能的微调提供了相关工具。
[0201]
蛋白质调查.测试了四批人血清白蛋白(hsa):hsa faf(不含脂肪酸)、hsa lfp(低叶酸粉末)、hsa rgp(试剂级粉末)和rhsa(重组hsa,来自大米)。此外,还测试了两种球蛋白,γ-球蛋白(人类来源)和血红蛋白(人类来源)。蛋白质的溶解度是albupad技术适用性的主要标准,选择这些球状蛋白质是因为它们具有足够的水溶性。每种蛋白质都使用以下盐进行测试:nabr、nacl和cacl2(盐/蛋白质的摩尔比:0
–
2000)。将蛋白质和盐的溶液在烘箱中于37℃蒸发7天。然后将干燥的材料洗涤并在水中浸泡48小时以去除盐分并选择水不溶性材料。用所有含有nabr和cacl2的人白蛋白获得水不溶性膜。这些膜稳定且易于处理。γ-球蛋白类膜用nabr适当地形成。因此,albupad技术可用于生产具有人血清白蛋白和重组人白蛋白以及人γ-球蛋白的稳定且易于处理的材料。
[0202]
2.2活性物质的加载
[0203]
由于白蛋白固有的加载各种物质的能力,基于白蛋白的材料有望用于药物递送和控释。此外,albupad技术允许在初始溶液中的材料形成之前加载活性物质(预加载),以及在材料形成之后将活性物质加载到材料中(后加载)。初步调查显示,albupad材料可以预加载亲脂性物质,如吡罗昔康和氟替卡松,以及水溶性分子洗必泰。在这项调查中,形成了稳定且易于处理的材料,从而验证了这种加载策略对albupad技术的适用性。此外,albupad材料中还加载了抗肿瘤药物多柔比星(dox)和用于治疗糖尿病的肽胰岛素(ins),以建立概念验证并研究这种物质的随时间的释放。通过荧光成像评估这些物质的加载。水中的药物释放定量通过荧光滴定随时间进行。
[0204]
预加载在albupad膜中的dox的加载和释放.bsa/nabr和bsa/cacl2膜是通过在蒸发前将dox直接加入溶液中预加载dox,如图21所示。溶液中加入不同量的dox,如下所示:每膜0.25、0.5、0.75和1mg dox(膜质量≈400mg)。在溶液蒸发后获得可处理的膜。由于dox的存在,制备的膜呈现颜色。此外,膜的着色强度与加载的dox的量成正比。为了可视化膜内dox的存在,进行clsm成像(图22)。在bsa/nabr膜中,dox的荧光均匀分布在膜基质内(厚度=500μm厚度),表明活性物质已成功加载到材料中(图22)。至于bsa/cacl2膜,材料的不透明性阻碍了观察加载在材料基质内的dox。然而,在它们的表面上观察到dox荧光,表明dox在这些膜中的加载也是成功的(图22)。
[0205]
接下来,将膜在水中洗涤以去除盐。将bsa/nabr膜在水(3x20ml)中洗涤2小时。将bsa/cacl2膜在水(4x20ml)中洗涤2小时。收集冲洗溶液并通过485nm的荧光光谱测量去除
的dox的量(图23)。对于bsa/nabr膜,去除的dox在20%到30%之间,对于bsa/cacl2膜,在35%到50%之间。因此,最初加载的dox的50%到70%在洗涤后仍然存在于膜中。
[0206]
在搅拌下,在37℃的水中研究dox从膜的释放35天。超过35天的上清液中的dox滴定显示,这种活性物质从albupad材料中的释放曲线缓慢且受控,具有有限的爆发效应(图24)。此外,根据制剂的不同,10%到50%的可用dox在35天后仍未释放,这表明该材料具有用于药物递送的潜力。
[0207]
预加载在albupad膜中的ins-fitc的加载和释放.
[0208]
bsa/nabr和bsa/cacl2膜是通过在蒸发前将ins-fitc直接加入溶液中预加载dox。溶液中加入的ins-fitc的量为每膜0.25mg(膜质量≈400mg)。在溶液蒸发后获得可处理的膜。为了可视化膜内ins-fitc的存在,进行clsm成像。在bsa/nabr膜中,ins-fitc的荧光均匀分布在膜基质内(厚度=500μm厚度),表明活性物质已成功加载到材料中(图25)。至于bsa/cacl2膜,材料的不透明性阻碍了观察加载在材料基质内的ins-fitc。然而,在它们的表面上观察到荧光,表明dox在这些膜中的加载也是成功的(图25)。
[0209]
接下来,将膜在水中洗涤以去除盐。将bsa/nabr膜在水(3x 20ml)中洗涤2小时。将bsa/cacl2膜在水(4x 20ml)中洗涤2小时。收集冲洗溶液并通过495nm的荧光光谱测量去除的ins-fitc的量(图27中a和b)。对于bsa/nabr和bsa/cacl2膜,在洗涤过程中分别去除了8%和1%的加载ins-fitc。因此,超过90%的ins-fitc在洗涤过程后在膜中可用。随后,在搅拌下,在37℃的水中研究ins-fitc从膜的释放30天。根据dox获得的结果,超过30天的上清液中的ins-fitc滴定显示,这种活性物质从albupad材料中的释放曲线缓慢且受控,具有有限的爆发效应(图26中c)。此外,根据制剂的不同,60%到80%的可用ins-fitc在30天后仍未释放,这表明该材料具有延长该活性物质释放的潜力。
[0210]
总之,考虑到所有进行的实验,证明了使用预加载方法加载材料的可行性。此外,庆大霉素等其他活性物质也成功加载在albupad膜中,加载率高达30%(wt/wt)。此外,最大加载速率取决于活性物质的溶解度及其与白蛋白的相互作用。因此,进一步的研究应该允许研究和优化目标活性物质的加载。此外,释放介质中蛋白酶的存在应该允许增加从膜中释放的物质。
[0211]
2.3生物相容性和体内生物降解性的初步评价
[0212]
使用电子束辐照优化albupad材料的灭菌.在将albupad材料植入体内之前,对使用电子束照射的灭菌进行了测试,以验证其与材料的相容性,从而不会导致其降解或其性质或组成它的白蛋白结构的显著变化。因此,制备了两种类型的膜:bsa/nabr和bsa/cacl2。这些样品的辐照被分成几组,以便在以下条件下测试它们在辐照下的稳定性:在存在或不存在辐射防护剂(维生素c)的情况下以及以干燥或水合(在水中)膜的形式。测试了四种辐射剂量:5、10、25和25kgy。将辐照膜的物理方面、质量损失和吸水量与未辐照的对照批次进行比较。此外,将辐照膜的ir光谱与对照的红外光谱进行比较,以检测白蛋白二级结构的变化。这些实验表明,在所有测试的辐照剂量下,材料的辐照都不会导致其降解,并且没有必要使用维生素c作为辐射防护剂。辐照样品与对照批次之间的比较表明,辐照后材料的性能没有显著变化。此外,ft-ir分析表明,材料的辐照对酰胺i带没有改变,因此不会破坏干燥样品和水合样品中白蛋白的二级结构。总之,电子束辐照灭菌与albupad材料兼容,并且可以在25kgy下有效地用于在这些材料进行体内评估之前对其进行灭菌。
[0213]
植入物配制、灭菌和小鼠皮下植入.为了对albupad材料的体内生物降解性和生物相容性进行初步研究,制备了以下批次的圆柱形植入物:bsa/nabr(bsa初始浓度=300mg/ml,nabr初始浓度=1.8m,n=5),bsa/cacl2(bsa的初始浓度=200mg/ml,cacl2的初始浓度=2.1m,n=10),hsa/nabr(hsa的初始浓度=300mg/ml,nabr的初始浓度=1.8m,n=5)和hsa/cacl2(hsa的初始浓度=200mg/ml,cacl2的初始浓度=2.1m,n=10)。通过在戊二醛存在下交联hsa(hsa/glu,n=5)制备了一批对照植入物。然后彻底清洗植入物并切割以适合推荐的尺寸(长度≈1cm,直径≤5mm)(图27中a)。这些材料的吸水率用于比较用bsa和hsa制备的批次。bsa/nabr和hsa/nabr具有相似的视觉方面和吸水值(分别为124
±
3%和127
±
21%),以及bsa/cacl2和hsa/cacl2(分别为224
±
52%和220
±
10%)(图27中b)。然后将植入物在24孔板中在37℃下干燥24小时。然后使用选定的方法(电子束,25kgy)对干燥批次的植入物进行灭菌。
[0214]
接下来,将无菌植入物用于小鼠皮下植入。将植入物在4℃下在pbs中再水化6小时,然后在植入前用pbs冲洗。对瑞士小鼠植入bsa/cacl2、hsa/cacl2和hsa/glu植入物(每种植入物三只小鼠)。对裸鼠植入bsa/nabr、bsa/cacl2、hsa/nabr和hsa/cacl2植入物。在植入bsa/glu材料的对照组中观察到急性毒性并导致该组小鼠被处死。植入albupad材料的小鼠在整个实验过程中(28天)保持活力,体重没有显著变化(
±5–
10%体重变化)。使用卡尺通过它们的皮肤测量植入物的体积。在瑞士小鼠组中,bsa/cacl2植入物在植入17天后完全降解;与此同时,hsa/cacl2植入物在28天后的实验结束时并未完全降解,平均损失了50%的体积。在裸鼠中,bsa/nabr、hsa/nabr、bsa/cacl2和hsa/cacl2植入物在植入28天后分别损失了23%、35%、53%和43%的体积。在第28天处死小鼠后,在gomori和picro sinius染色后通过显微镜对植入物及其周围组织的切口进行组织学评估。对准备好的切口的观察表明,植入物没有碎裂,表明它们的生物降解是渐进的,不会导致大碎片的形成(图28中a)。还观察到植入物周围的组织表型是正常的,并且胶原纤维的形成和取向与在天然组织中观察到的相似(图28中b)。在与材料接触时未观察到纤维增殖。因此,测试的植入物对周围组织或小鼠是无毒的,并且被证明在体内是可生物降解的,其速率取决于植入物的类型。
[0215]
实验部分
[0216]
化学试剂.牛血清白蛋白(部分v,≥96%)购自acros organics。从sigma-aldrich购买血红蛋白(人)、牛血中的γ-球蛋白(≥99%)、重组人白蛋白(rhsa,在大米中表达)、溴化钠(nabr)、氯化钾(kcl)、多柔比星(dox)、与fitc结合的胰岛素(ins-fitc)和氧化氘(d2o)。氯化钠(nacl)购自vwr chemicals。氯化镁(mgcl2,无水)购自fluka。氯化钙(cacl2,2h2o)购自merck。从sera care购买了以下人血清白蛋白(hsa):hsa试剂级粉末(rgp)、hsa低叶酸粉末(lfp)和hsa无脂肪酸(faf)。
[0217]
制剂(一般程序).制备bsa(100mg/ml)和nabr(1m)在乙酸钠缓冲液(0.2m)中的ph 6溶液。将该溶液沉积在硅树脂模具中并在37℃下蒸发直至干燥。将获得的干燥生物材料洗涤以除去盐,并在室温下在蒸馏水中浸泡48小时。然后收集和表征水不溶性膜(bsa/nabr)。
[0218]
初始表征.使用相对产率(式s1)、吸水率(式s2)和初始膨胀(式s3)比较配制的膜。wbsa代表用于制剂的白蛋白的初始质量。ai代表在蒸发过程中使用的圆形容器的面积。wd表示在蒸馏水中洗涤48小时并在37℃烘箱中干燥过夜后最终干燥膜的质量。vd是通过将材料在室温下浸入蒸馏水中测量的干燥膜的体积。wh表示在蒸馏水中浸泡24小时并使用滤纸去
除多余的表面水后处于平衡状态的水合膜的质量。ah是浸入蒸馏水中24小时后处于平衡状态的水合膜表面的面积,使用电子数显卡尺(tacklife-dc01,精度
±
0.2mm)测量其直径后计算得出。
[0219][0220][0221][0222]
扫描电子显微镜(sem).使用quanta
tm 250feg sem(fei company,eindhoven,the netherlands)进行扫描电子显微镜和微量分析评估,在电子加速电压10kv下操作。对于sem实验,将bsa/nabr 664膜干燥,然后使用hummer jr溅射装置(technics,union city,ca,usa)涂上金钯合金。
[0223]
在有机溶剂中稳定性.将bsa/nabr膜置于5ml以下有机溶剂中:dmso、乙腈和二氯甲烷。然后将含有膜的培养基在室温下伴随搅拌培养72小时。之后,在表征其质量损失(式s4)之前用水洗涤膜。
[0224][0225]
膜的机械性能.使用平行板配置(直径20mm)的流变仪kinexus ultra (malvern,英国)进行压缩测试。使用一批三水合(在蒸馏水中)bsa/nabr膜(厚度=0.7mm)。弹性模量(e)在应变(ε)-应力(σ)曲线(式s5)的弹性域内计算。
[0226]
σ=exε
ꢀꢀꢀꢀ
(s5)
[0227]
流变测量.使用平行板配置(直径20mm)的流变仪kinexus ultra (malvern,英国)评估bsa/nabr膜(厚度=0.7mm)的粘弹性行为。膜预先在蒸馏水中水合24小时。将bsa/nabr圆片加载到板之间,并封闭间隙直到样品与两个板良好接触(法向力《1n)。在实验开始以及实验之间,将样品平衡5分钟。在此期间,所有样品的法向力均降至0.1n以下。幅度扫描测试是在0.01-100%的剪切应变下以0.5hz的固定频率在25℃下进行的。剪切模量g*(ω)=σ(ω)/γ(ω)来自应力(σ)和应变幅值(γ)之间的比率。g*(ω)=g' ig”是弹性储能模量(g')和粘性损耗模量(g”)的复数。使用|g*|=(g
’2 g”2
)
0.5
计算剪切模量的绝对值|g*|。
[0228]
接触角测量.使用了来自biolin scientific(瑞典)的theta张力计和oneattension软件。实验以“静滴”模式进行,以测量水在干燥的bsa/nabr膜上的静态接触角。使用hamilton微量注射器(针头直径=0.7mm)分配水滴。每次测量均使用5μl的milliq水滴(滴速=2μl/s)进行,每个样品重复三次。内置摄像头捕获了静滴的一系列图像(帧速率=14fps),同时测量水滴与材料相交处水滴形成的切线角。10秒后记录平均接触角(左右接触角的平均值),该时间足以使液滴稳定。
[0229]
ir分析.ftir实验在vertex 70光谱仪(德国bruker)上使用dtgs检测器进行。使用blackman-harris三项切趾法和bruker opus/ir软件(7.5版),通过在800和4000cm-1
之间以2cm-1
分辨率平均128个干涉图,采用单反射金刚石atr,以衰减全反射(atr)模式记录光谱。将现成的bsa/nabr膜(干燥)精细研磨并记录光谱。为了分解酰胺i带区域(1700
–
1600cm-1
),使用opus 7.5软件(bruker optik gmbh)进行数据处理。在曲线拟合之前,对酰胺i带区域的光谱进行基线校正,然后使用归一化“最小-最大”方法进行归一化。子带的数量及其位置由光谱的四阶导数确定。根据最小二乘迭代曲线拟合程序(levenberg-marquardt)使用高斯线形进行去卷积。在最后的拟合中,为了尽可能减少残余rms误差(小于0.005),调整了所有带的高度、宽度和位置,同时每次都不允许更改这些参数中的至少一个。最后,比较原始曲线和拟合曲线的二阶导数,保证曲线拟合的准确性。在根据文献识别它们后,拟合分量的分数面积用于计算不同二级结构元素(α螺旋、β折叠、β转角和无规卷曲)的百分比。
[0230]
预加载制剂.bsa溶液(100mg/ml)在ph 6的醋酸盐缓冲液(0.2m)中制备,并与nabr(nabr/bsa摩尔比=400)或cacl2(cacl2/bsa摩尔比=700)盐混合。对于多柔比星(dox)的预加载,在蒸发前将以下量的dox添加到初始溶液中:每膜0.25、0.5、0.75和1mg(膜质量=400mg)。对于胰岛素(ins-fitc)的预加载,在蒸发前将每膜0.25mg ins-fitc(膜质量=400mg)添加到初始溶液中。将溶液沉积在防粘硅胶模具中,并在37℃下蒸发48小时。获得的干生物材料在对于bsa/nabr膜为3x 20ml水中和对于bsa/cacl2膜为4x 20ml水中彻底洗涤,以去除盐分。
[0231]
共焦激光扫描显微镜表征(clsm).为了可视化预加载膜内dox或ins-fitc的存在,使用zeiss lsm 710共聚焦显微镜进行clsm成像。对于dox和ins-fitc预加载材料,膜成像的激发/发射波长固定为450/515nm。
[0232]
释放实验.dox或ins-fitc预加载膜的释放用genius xc荧光分光计(safas,摩纳哥)监测。释放实验在37℃的h2o(每个膜10ml)中进行。每次收集上清液后加入新鲜的10ml水。用荧光分光计分析上清液。dox和ins-fitc的激发和发射波长分别为λ
ex
/λ
em
=485nm/595nm和λ
ex
/λ
em
=495nm/520nm。
[0233]
统计分析.使用r(3.6.1版,r foundation for statistical computing,vienna,austria)分析数据。分布的正态性由shapiro-wilk检验确定。用f检验确定方差的相等性。当数据呈正态分布且样本方差相等时,使用t检验(2尾)比较两个均值。当这两个条件不适用时,改为执行mann-whitney检验。在p《0.05时值被认为具有统计学意义。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。