1.本发明涉及生物基因工程领域,尤其涉及一种基于全基因组数据检测可移动遗传元件的方法。
背景技术:
2.可移动遗传元件是一类可在细菌胞内或胞间移动的dna片段,能够编码一种或多种影响毒力或抗性传播因素,包括介导自身转移和整合的酶。可移动遗传元件包括质粒、插入序列、原噬菌体、整合子和转座子等,它们彼此之间经常存在嵌套、插入、颠转、截短等多种关系,以形成更加复杂的结构。
3.中国专利公告号cn107922936a公开了识别影响感兴趣的细胞内表型的内源生理相关的遗传元件的通用方法,该方法将已进行诱变处理的非活细胞基于表型进行分选并分析从而识别遗传元件,可识别先前未知参与表型的元件。上述技术方案中新出现的可移动元件不能及时在数据库中更新和管理。
技术实现要素:
4.本发明主要解决原有技术中可移动遗传元件数据库缺乏有效和可持续的管理流程,致使新出现的可移动元件不能及时在数据库中更新和管理的问题,各可移动元件数据库的命名、分类不统一,提供一种基于全基因组数据检测可移动遗传元件的方法,利用mge特征序列提取算法获取可移动遗传元件特征序列,并通过blast工具得到可移动遗传元件预测集和结果集,并为可移动遗传元件数据库的命名提供统一的格式,防止同样的可移动遗传元件误认成不同可移动遗传元件。
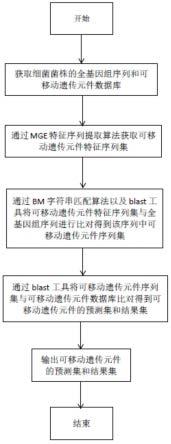
5.本发明的上述技术问题主要是通过下述技术方案得以解决的,本发明包括一种基于全基因组数据检测可移动遗传元件的方法,包括以下步骤:s1 获取细菌菌株的全基因组序列和可移动遗传元件数据库;s2 通过mge特征序列提取算法获取可移动遗传元件特征序列集;s3 通过bm字符串匹配算法以及blast工具将可移动遗传元件特征序列集与全基因组序列进行比对得到该序列中可移动遗传元件序列集;s4 将可移动遗传元件序列集与可移动遗传元件数据库通过blast工具比对得到可移动遗传元件的预测集和结果集;s5 输出可移动遗传元件的结果集,预测集用于实验验证。
6.利用mge特征序列提取算法获取可移动遗传元件特征序列集,再通过bm字符串匹配算法以及blast工具获取可移动遗传元件序列集,并通过blast工具得到可移动遗传元件预测集和结果集,结果集用于对全基因组测序数据直接注释,预测集用于后续实验验证,验证为可移动遗传元件则补充至数据库,有效解决了可移动遗传元件数据库无法及时更新,导致无法检查出全部的可移动遗传元件的问题,进一步提高了可移动移传元件的检出率。
7.作为优选,所述全基因组序列具体从ncbi数据库获取数据,并以fasta格式进行存
储。
8.数据库权威性高,获得数据的准确性高。作为优选,所述可移动遗传元件数据库包括插入序列数据库、整合子数据库和转座子数据库,所述可移动遗传元件数据库分别从isfinder数据库、integrall数据库和the transpson registry数据库中获取插入序列数据库、整合子数据库和转座子数据库,并以fasta格式进行存储并通过blast工具格式化成本地库。针对不同的可移动遗传元件采用专门的权威数据库,获得数据的准确性高,blast工具比对是一种在大量序列中寻找和待查询序列具有局部相似性的片段的近似算法,准确性高,覆盖范围广。
9.作为优选,所述可移动遗传元件特征序列集包括插入序列末端反向重复序列集、整合酶的基因序列集、整合酶附着位点序列集和转座子正向重复序列集,所述插入序列末端反向重复序列集通过mge特征序列提取算法在插入序列数据库中获取,所述整合酶的基因序列集和整合酶附着位点序列集通过mge特征序列提取算法在整合子数据库中获取,所述转座子正向重复序列集通过mge特征序列提取算法在转座子数据库中获取。
10.mge特征序列提取算法包含特征序列提取、特征序列去冗余和特征序列集本地化;输入插入序列数据库,根据基因水平结构,对插入序列进行末端反复序列提取,长度为10到40bp,并将提取序列去除冗余部分,插入序列末端反向重复序列集保存为fasta格式并格式化成本地库;通过整合子数据库收集整合酶的基因序列集及整合酶附着位点序列集保存为fasta格式并分别格式化成本地库;输入转座子数据库,可根据基因水平结构,对转座子进行正向序列提取,长度为5到9个bp,并将提取序列去除冗余部分,将转座子正向重复序列集保存为fasta格式并格式化成本地库。
11.作为优选,所述插入序列末端反向重复序列集的格式具体为插入序列名称,末端重复序列左侧,末端重复序列右侧;所述整合酶的基因序列集及整合酶附着位点序列集的格式具体为ncbi序列登录号,基因,序列,整合酶附着位点序列;所述转座子正向重复序列集的格式具体为转座子名称,正向重复序列。为可移动元件数据库的命名提供统一的格式,防止同样的可移动遗传元件误认成不同可移动遗传元件。
12.作为优选,所述可移动遗传元件序列集包括插入序列集、转座子集、整合酶基因及其附着位点集,所述插入序列集具体为插入序列末端反向重复序列集通过字符串匹配算法与全基因组序列比对获得,所述转座子集具体为转座子正向重复序列集通过字符串匹配算法与全基因组序列比对获得,所述整合酶基因及其附着位点集具体为整合酶的基因序列集和整合酶附着位点序列集通过blast工具与全基因组序列比对获得。
13.字符串匹配算法可以减少匹配所耗费的时间,能够提高总体性能,通过字符串匹配算法,从全基因组序列中找到成对插入序列末端反向重复序列后将起始点与终止点之间的序列截取以fasta格式保存,得到插入序列集;通过字符串匹配算法,从全基因组序列中找到成对转座子两端正向重复序列后将起始点与终止点之间的序列截取以fasta格式保存,得到转座子集;通过blast工具,从全基因组序列中找到整合酶基因序列与整合酶结合位点序列后将起始点与终止点之间的序列截取以fasta格式保存,得到整合酶基因及其附着位点集。
14.作为优选,所述插入序列集的格式具体为插入序列序号,起始点,终止点,末端反向重复序列起始点与终止点,截取的序列信息;所述转座子集的格式具体为转座子序号,起
始点,终止点,正向重复序列起始点与终止点,截取的序列信息;所述整合酶基因及其附着位点集的格式具体为整合酶基因序列,起始点,终止点,整合酶附着位点起始点与终止点以及序列信息。提供统一的格式,防止同样的可移动遗传元件误认成不同可移动遗传元件。
15.作为优选,所述预测集包括插入序列预测集和转座子预测集,所述结果集包括插入序列结果集、转座子结果集和整合酶基因及其附着位点结果集,所述插入序列结果集和插入序列预测集具体为插入序列集通过blast工具与插入序列数据库进行比对获得,所述转座子结果集和转座子预测集具体为转座子集通过blast工具与转座子数据库进行比对获得,所述整合酶基因及其附着位点结果集具体为整合酶基因及其附着位点集通过blast工具与整合子数据库进行比对获得。结果集用于对全基因组测序数据直接注释,预测集用于后续实验验证,验证为可移动遗传元件则补充至数据库。
16.本发明的有益效果是:本发明提供一种基于全基因组数据检测可移动遗传元件的方法,利用mge特征序列提取算法获取可移动遗传元件特征序列,并通过blast工具得到可移动遗传元件预测集和结果集,采用全基因组测序技术对未知基因组序列的细菌进行个体分析测定完整的基因序列信息,有效解决了可移动遗传元件数据库无法及时更新,导致无法检查出全部的可移动遗传元件的问题,进一步提高了可移动移传元件的检出率,并为可移动遗传元件数据库的命名提供统一的格式,防止同样的可移动遗传元件误认成不同可移动遗传元件。
附图说明
17.图1是本发明的一种逻辑流程示意图。
18.图2是本发明的一种工作方法示意图。
19.图3是本发明的一种mge特征序列提取算法流程图。
具体实施方式
20.下面通过实施例,并结合附图,对本发明的技术方案作进一步具体的说明。
21.实施例:一种基于全基因组数据检测可移动遗传元件的方法,下面以鲍曼不动杆菌全基因组序列为例,包括下列步骤:s1通过ncbi数据库中获取已完成拼接的鲍曼不动杆菌全基因组序列,从isfinder数据库中获取插入序列数据库,从integrall数据库中获取整合子数据库,从the transpson registry数据库中获取转座子数据库;s2 通过mge特征序列提取算法获取可移动遗传元件特征序列集;输入插入序列数据库,包括5550个插入序列,属性包括插入序列名称,来源菌属通过mge特征序列算法根据插入序列末端反复序列长度进行截取,长度为10-40bp,属性保留,去除冗余部分,获得插入序列末端反复序列集,通过blast工具进行本地化制备成本地库;输入整合子数据库,包括11957个整合子序列,属性包括整合子基因来源菌属,整合子ncbi登录号,基因盒包含基因名称,从中获取整合酶基因序列集及其附着位点序列集,属性保留,同样去除冗余部分后通过blast格式化为本地库;输入转座子数据库,包含1679个转座子,属性包括名称,ncbi登录号,来源菌属,从中获取转座子两端正向重复序列集,长度为5-9 bp,属性保留,同样需要去除冗余部分后通过blast格式化为本地库;
s3 通过bm字符串匹配算法以及blast工具将可移动遗传元件特征序列集与全基因组序列进行比对得到该序列中可移动遗传元件序列集,所述可移动遗传元件序列集包括插入序列集、转座子集、整合酶基因及其附着位点集;插入序列末端反复序列集中序列相对长度较短,并且序列以字符串形式保存,可通过字符串匹配算法将以上集合中的短序列与输入的鲍曼不动杆菌全基因组序列进行匹配得到可能的插入序列集; 转座子两端正向重复序列集相对长度较短,并且序列以字符串形式保存,可通过字符串匹配算法将以上集合中的短序列与输入的鲍曼不动杆菌全基因组序列进行匹配得到可能的转座子集;使用的是bm字符串匹配算法,匹配结果输出结果包括起始位置、长度以及步骤s2中保留的属性;整合酶基因序列相对较长,可通过blast工具与鲍曼不动杆菌全基因组序列进行比对,最小覆盖率与一致性一般为60%和90%,输出结果包括基因序列,起始位置,长度,及步骤s2中保留属性;s4 将步骤s3得到的可移动遗传元件序列集与步骤s1中数据库分别通过blast进行比对,得到可移动遗传元件预测集和结果集。
22.s5 输出可移动遗传元件的预测集和结果集,结果集用于对全基因组测序数据直接注释,预测集用于后续实验验证,验证为可移动遗传元件则补充至数据库。
23.本文中所描述的具体实施例仅仅是对本发明精神作举例说明。本发明所属技术领域的技术人员可以对所描述的具体实施例做各种各样的修改或补充或采用类似的方式替代,但并不会偏离本发明的精神或者超越所附权利要求书所定义的范围。
24.尽管本文较多地使用了可移动遗传元件、全基因组序列等术语,但并不排除使用其它术语的可能性。使用这些术语仅仅是为了更方便地描述和解释本发明的本质;把它们解释成任何一种附加的限制都是与本发明精神相违背的。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。