含硅的抗蚀剂下层膜形成用组成物以及图案形成方法
【技术领域】
1.本发明关于含硅的抗蚀剂下层膜形成用组成物、及使用该组成物的图案形成方法。
背景技术:
2.伴随大型集成电路(lsi)的高集成化与高速化,图案尺寸的微细化正急速进展。光刻技术,伴随该微细化,通过光源的短波长化及适当选择与其对应的抗蚀剂组成物,已达成微细图案的形成。成为其中心的是以单层使用的正型光致抗蚀剂组成物。该单层正型光致抗蚀剂组成物,通过使抗蚀剂树脂中具有对于利用氯系或氟系气体等离子所为的干蚀刻具蚀刻耐性的骨架,且具有曝光部会溶解之类的开关(switching)机制,而使曝光部溶解来形成图案,并将残存的抗蚀剂图案作为蚀刻掩膜对被加工基板进行干蚀刻加工。
3.然而,于维持所使用的光致抗蚀剂膜的膜厚的状态进行微细化,也即将图案宽进一步缩小时,光致抗蚀剂膜的分辨性能降低,又,欲利用显影液对光致抗蚀剂膜进行图案显影的话,会发生所谓纵横比变得过大,结果造成图案崩坏的问题。因此,伴随图案的微细化,光致抗蚀剂膜也逐渐薄膜化。
4.另一方面,被加工基板的加工,通常使用将形成有图案的光致抗蚀剂膜作为蚀刻掩膜,并利用干蚀刻对基板进行加工的方法,但现实中并不存在能于光致抗蚀剂膜与被加工基板之间取得完全的蚀刻选择性的干蚀刻方法。因此,会有在基板的加工中,抗蚀剂膜也受到损伤而崩坏,且抗蚀剂图案变得无法正确地转印至被加工基板的问题。因此,伴随图案的微细化,光致抗蚀剂组成物也逐渐寻求更高的干蚀刻耐性。但是,另一方面,为了提高分辨率,光致抗蚀剂组成物所使用的树脂已逐渐寻求在曝光波长的光吸收小的树脂。因此,随着曝光光短波长化为i射线、krf、arf,光致抗蚀剂组成物所使用的树脂也逐渐变化成酚醛清漆树脂、具有多羟基苯乙烯、脂肪族多环状骨架的树脂,但现实中基板加工时的干蚀刻条件下的蚀刻速度逐渐变快,分辨率高的最近的光致抗蚀剂组成物反而会有蚀刻耐性变弱的倾向。
5.考量此情形,变得必须以较薄且蚀刻耐性较弱的光致抗蚀剂膜对被加工基板进行干蚀刻加工,确保该加工步骤中的材料及处理已成为当务之急。
6.作为解决如此的问题的方法之一,有多层抗蚀剂法。该方法是将蚀刻选择性与光致抗蚀剂膜(也即,抗蚀剂上层膜)不同的下层膜插入在抗蚀剂上层膜与被加工基板之间,在抗蚀剂上层膜获得图案后,将抗蚀剂上层膜图案作为干蚀刻掩膜,利用干蚀刻将图案转印至下层膜,进一步,将下层膜作为干蚀刻掩膜,利用干蚀刻将图案转印至被加工基板的方法。
7.作为多层抗蚀剂法之一,有可使用单层抗蚀剂法中所使用的一般的抗蚀剂组成物来实施的3层抗蚀剂法。该3层抗蚀剂法中,例如于被加工基板上将酚醛清漆树脂等构成的有机膜予以成膜,于其上将含硅的抗蚀剂下层膜予以成膜,再于上形成通常的有机系光致抗蚀剂膜作为抗蚀剂上层膜。实施利用氟系气体等离子所为的干蚀刻时,有机系抗蚀剂上
层膜相对于含硅的抗蚀剂下层膜可取得良好的蚀刻选择比,故抗蚀剂上层膜图案可利用氟系气体等离子所为的干蚀刻而转印至含硅的抗蚀剂下层膜。根据该方法,即使使用难以形成具有用以直接对被加工基板进行加工所需的充分膜厚的图案的抗蚀剂组成物、不具有基板的加工所需的充分干蚀刻耐性的抗蚀剂组成物,也可将图案转印至含硅膜(抗蚀剂下层膜),然后利用氧系或氢系气体等离子所为的干蚀刻实施图案转印,则可获得具有基板的加工所需的充分干蚀刻耐性的酚醛清漆树脂等构成的有机膜的图案。形成有机膜图案后所残留的含硅的抗蚀剂下层膜,通常利用氟系气体等离子所为的干蚀刻、或碱系、氟系等蚀刻液所为的湿式蚀刻,以不成为造成缺陷的原因的残渣的方式予以去除。蚀刻速度不足时,发生来自含硅的抗蚀剂下层膜的残渣残留而成为缺陷、或需要进行长时间的蚀刻处理而对被加工基板造成损伤等问题的可能性提高。如此,为了正确的图案化、及顺利的去除,含硅的抗蚀剂下层膜需要适当的蚀刻速度。
8.另一方面,近年由于arf浸润式光刻、euv光刻等的出现,逐渐可形成更微细的图案,但另一方面,超微细图案由于接地面积小,故极易崩塌,图案形状的改善与图案崩塌的抑制是非常大的课题。因此,抑制图案崩塌的效果高的含硅的抗蚀剂下层膜的开发成为当务之急。
9.为了改善图案崩塌,有人提出于硅氧烷结构的侧链结构导入了具有密接性、热硬化催化剂功能等效果的取代基的arf或euv光刻用含硅的抗蚀剂下层膜形成用组成物(专利文献1、2)。通过它们的导入而抑制图案崩塌的效果得到改善,但在使用了euv光刻等的更进一步微细化的最先进的用途中,图案崩塌依然是重大的课题。
10.[现有技术文献]
[0011]
[专利文献]
[0012]
[专利文献1]wo2020/085508号
[0013]
[专利文献2]wo2020/196563号
技术实现要素:
[0014]
[发明所欲解决的课题]
[0015]
本发明旨在提供于多层抗蚀剂法中,抑制超微细图案崩塌的效果高,可形成图案形状良好的抗蚀剂图案的含硅的抗蚀剂下层膜。
[0016]
[解决课题的手段]
[0017]
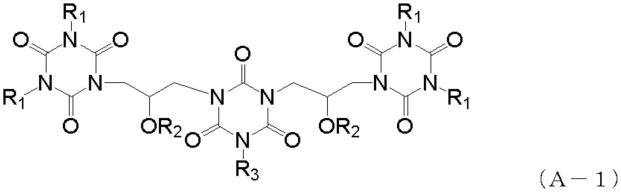
为了解决上述课题,本发明提供一种含硅的抗蚀剂下层膜形成用组成物,含有下列通式(a-1)表示的化合物及热交联性聚硅氧烷。
[0018]
[化1]
[0019][0020]
r1表示甲基、乙基、丙基、烯丙基、炔丙基,r2表示氢原子、乙酰基、丙烯酰基、甲基丙
烯酰基、苯甲酰基、萘甲酰基、蒽甲酰基,r3表示甲基、乙基、丙基、烯丙基、炔丙基、或下列通式(a-2)表示的基团。
[0021]
[化2]
[0022][0023]
虚线表示原子键,r1、r2与前述相同。
[0024]
在半导体装置制程中使用的使用了arf或euv等紫外线激光的光刻步骤中,通过使用由含有具有通式(a-1)表示的异氰尿酸结构的化合物的本发明的组成物形成的含硅的抗蚀剂下层膜,可对形成于其上的光致抗蚀剂图案赋予良好的密接性。
[0025]
前述热交联性聚硅氧烷宜含有下列通式(sx-1)表示的重复单元、下列通式(sx-2)表示的重复单元、及下列通式(sx-3)表示的部分结构中的任一者以上。
[0026]
[化3]
[0027][0028]
[化4]
[0029][0030]
[化5]
[0031][0032]
式中,r4、r5、r6可各自相同也可不同地为碳数1~30的1价有机基团。
[0033]
上述热交联性聚硅氧烷(sx)含有上述特定结构时,会更充分地发挥本发明的效果。
[0034]
宜更含有酸产生剂。
[0035]
通过视需要添加酸产生剂,可对图案形状、曝光感度等进行微调整。
[0036]
前述酸产生剂宜为是锍盐且会因高能量射线的作用而产生酸的光酸产生剂较佳。
[0037]
此时,可将其他性能的降低压抑为最小限度,同时可适度调整抗蚀剂上层膜的图案形状、曝光感度等,且有时于减少来自抗蚀剂上层膜的残渣也是有效的。
[0038]
又,本发明提供一种图案形成方法,是于被加工体形成图案的方法,包含下列步骤:
[0039]
于被加工体上使用涂布型有机膜材料来形成有机膜;
[0040]
于前述有机膜上使用上述含硅的抗蚀剂下层膜形成用组成物来形成含硅的抗蚀剂下层膜;
[0041]
于前述含硅的抗蚀剂下层膜上使用由光致抗蚀剂组成物构成的抗蚀剂上层膜用组成物来形成抗蚀剂上层膜;
[0042]
于前述抗蚀剂上层膜形成电路图案;
[0043]
将形成有前述电路图案的前述抗蚀剂上层膜作为掩膜,利用蚀刻将图案转印至前述含硅的抗蚀剂下层膜;
[0044]
将转印有前述图案的前述含硅的抗蚀剂下层膜作为掩膜,利用蚀刻对前述有机膜进行图案转印;及
[0045]
将转印有前述图案的前述有机膜作为掩膜,利用蚀刻将图案转印至前述被加工体。
[0046]
又,本发明提供一种图案形成方法,是于被加工体形成图案的方法,包含下列步骤:
[0047]
于被加工体上利用cvd法形成以碳作为主成分的硬掩膜;
[0048]
于前述硬掩膜上使用上述含硅的抗蚀剂下层膜形成用组成物来形成含硅的抗蚀剂下层膜;
[0049]
于前述含硅的抗蚀剂下层膜上使用由光致抗蚀剂组成物构成的抗蚀剂上层膜用组成物来形成抗蚀剂上层膜;
[0050]
于前述抗蚀剂上层膜形成电路图案;
[0051]
将前述形成有电路图案的抗蚀剂上层膜作为掩膜,利用蚀刻将图案转印至前述含硅的抗蚀剂下层膜;
[0052]
将转印有前述图案的前述含硅的抗蚀剂下层膜作为掩膜,利用干蚀刻对前述硬掩膜进行图案转印;及
[0053]
将转印有前述图案的前述硬掩膜作为掩膜,利用干蚀刻将图案转印至前述被加工体。
[0054]
上述图案形成方法可抑制抗蚀剂上层膜的图案崩塌,且可提供良好的抗蚀剂图案。又,利用干蚀刻所为的朝含硅的抗蚀剂下层膜的图案转印优异,且图案化结束后所残存的含硅的抗蚀剂下层膜的去除容易,不易产生因残渣所致的缺陷,故尤其于微细图案的形成是实用的图案形成方法。
[0055]
于前述抗蚀剂上层膜形成电路图案的步骤中的图案形成,宜利用波长为10nm以上300nm以下的光学光刻、电子束所为的直接描绘、纳米压印或它们的组合来形成图案较佳。
[0056]
上述抗蚀剂上层膜的图案形成方法为上述特定的图案形成方法时,会更充分地发挥本发明的效果。
[0057]
前述被加工体宜为半导体装置基板、金属膜、金属碳化膜、金属氧化膜、金属氮化膜、金属氧化碳化膜或金属氧化氮化膜。
[0058]
被加工体为上述特定者时,会更充分地发挥本发明的效果。
[0059]
另外,构成前述被加工体的金属宜为硅、镓、钛、钨、铪、锆、铬、锗、铜、银、金、铟、砷、钯、钽、铱、铝、铁、钼、钴或它们的合金。
[0060]
构成被加工体的金属为上述特定的金属时,会更充分地发挥本发明的效果。
[0061]
[发明的效果]
[0062]
使用了含有具有多个异氰尿酸结构的化合物的本发明的含硅的抗蚀剂下层膜形成用组成物的含硅的抗蚀剂下层膜,可无崩塌地形成超微细的上层抗蚀剂图案,不仅如此,还可形成良好的图案形状。又,抗蚀剂上层膜与有机膜或与硬掩膜的干蚀刻选择性优异,故能以良好产能于基板形成半导体装置用图案。另外,使用了本发明的含硅的抗蚀剂下层膜形成用组成物的含硅的抗蚀剂下层膜,可在与有机材料之间获得高蚀刻选择性,故可将形成的光致抗蚀剂图案利用干蚀刻处理依序转印至含硅的抗蚀剂下层膜、有机膜或硬掩膜。又,本发明中,形成的含硅的抗蚀剂下层膜具有充分的蚀刻速度,故图案化结束后所残存的含硅的抗蚀剂下层膜的去除容易,不易产生因残渣所致的缺陷,因而于微细图案的形成尤其有用。
【具体实施方式】
[0063]
本发明关于用以形成半导体元件等的制造步骤中的光刻所使用的涂布型含硅膜的组成物及使用了该组成物的图案形成方法。
[0064]
如前述,寻求能提供于多层抗蚀剂法中,抑制超微细图案崩塌的效果高,且可形成良好的抗蚀剂图案,具有适当蚀刻速度的含硅的抗蚀剂下层膜的含硅的抗蚀剂下层膜形成用组成物、及使用了该组成物的超微细图案形成方法。
[0065]
本技术发明人等为了达成前述目的而进行努力研究的结果,发现通过将具有特定结构的异氰尿酸结构的化合物掺合至含硅的抗蚀剂下层膜形成用组成物中,能提供于多层抗蚀剂法中可抑制超微细图案的崩塌,且可形成良好的抗蚀剂图案,而且具有适合加工的蚀刻速度的含硅的抗蚀剂下层膜,而完成了本发明。
[0066]
也即,本发明是一种含硅的抗蚀剂下层膜形成用组成物,含有下列通式(a-1)表示的化合物及热交联性聚硅氧烷。
[0067]
[化6]
[0068][0069]
r1表示甲基、乙基、丙基、烯丙基、炔丙基,r2表示氢原子、乙酰基、丙烯酰基、甲基丙烯酰基、苯甲酰基、萘甲酰基、蒽甲酰基,r3表示甲基、乙基、丙基、烯丙基、炔丙基、或下列通式(a-2)表示的基团。
[0070]
[化7]
[0071][0072]
虚线表示原子键,r1、r2与前述相同。
[0073]
以下,针对本发明详细地说明,但本发明不限定于这些。
[0074]
《含硅的抗蚀剂下层膜形成用组成物》
[0075]
本发明的含硅的抗蚀剂下层膜形成用组成物,其特征为含有:下列通式(a-1)表示的化合物及热交联性聚硅氧烷。
[0076]
[化8]
[0077][0078]
r1表示甲基、乙基、丙基、烯丙基、炔丙基,r2表示氢原子、乙酰基、丙烯酰基、甲基丙烯酰基、苯甲酰基、萘甲酰基、蒽甲酰基,r3表示甲基、乙基、丙基、烯丙基、炔丙基、或下列通式(a-2)表示的基团。
[0079]
[化9]
[0080]
[0081]
虚线表示原子键,r1、r2与前述相同。
[0082]
[通式(a-1)表示的化合物]
[0083]
上述通式(a-1)表示的化合物可例示下列等。r3与前述相同。
[0084]
[化10]
[0085][0086]
[化11]
[0087][0088]
[化12]
[0089][0090]
[化13]
[0091][0092]
[化14]
[0093][0094]
上述通式(a-2)表示的结构可例示下列等。
[0095]
[化15]
[0096][0097]
[化16]
[0098][0099]
上述通式(a-1)表示的化合物的较佳例可例示下列等,具体而言,r1为烯丙基或炔丙基,r2为氢原子、乙酰基、丙烯酸基,r3为烯丙基或上述通式(a-2)表示的基团较佳,r1为烯丙基或炔丙基,r2为氢原子、乙酰基,r3为烯丙基尤佳。
[0100]
[化17]
[0101][0102]
[化18]
[0103][0104]
[通式(a-1)表示的化合物的合成方法]
[0105]
作为获得本发明中使用的通式(a-1)表示的化合物的手段,并无特别限定,可利用如下列反应式表示的环氧化合物与异氰尿酸衍生物的加成反应(step1-1)获得,r2不为氢原子时,可后续利用酰化反应(step1-2)获得。但是,r3为(a-2)时,可通过使用三官能团的环氧化合物作为环氧化合物并进行同样的反应(step2-1、step2-2)来获得。下式中的r1、r2、r3与前述相同。
[0106]
[化19]
[0107]
step1-1
[0108][0109]
[化20]
[0110]
step1-2
[0111][0112]
[化21]
[0113]
step2-1
[0114][0115]
[化22]
[0116]
step2-2
[0117][0118]
上述step1-1、或step2-1表示的环氧化合物与异氰尿酸衍生物的反应,令环氧化合物中的环氧量的摩尔量为1摩尔时,异氰尿酸衍生物的进料量宜为0.3~2.0摩尔,更佳0.5~1.5摩尔,尤佳为0.75~1.25摩尔。
[0119]
异氰尿酸衍生物的进料量相对于环氧单元只要没有不足,则不会有未反应的环氧基残存,且不会影响保存稳定性。异氰尿酸衍生物的进料量相对于环氧单元只要不过剩,则不会有未反应的异氰尿酸衍生物残存,且不会产生散逸气体。
[0120]
由如上述的原料合成的化合物,通常可通过在溶剂中或溶剂中且反应催化剂存在下,于室温或视需要冷却或加热下使环氧化合物与异氰尿酸衍生物反应而获得。
[0121]
所使用的溶剂可例示:甲醇、乙醇、异丙醇、丁醇、乙二醇、丙二醇、二乙二醇、甘油、甲基溶纤剂、乙基溶纤剂、丁基溶纤剂、丙二醇单甲醚等醇类;二乙醚、二丁醚、二乙二醇二乙醚、二乙二醇二甲醚、四氢呋喃、1,4-二噁烷等醚类;二氯甲烷、氯仿、二氯乙烷、三氯乙烯等氯系溶剂类;己烷、庚烷、苯、甲苯、二甲苯、异丙苯等烃类;乙腈等腈类;丙酮、甲乙酮、异
丁基甲基酮等酮类;乙酸乙酯、乙酸正丁酯、丙二醇甲醚乙酸酯等酯类;γ-丁内酯等内酯类;二甲基亚砜、n,n-二甲基甲酰胺、六甲基磷酰三胺等非质子性极性溶剂类,它们可单独使用或将2种以上混合使用。这些溶剂能以相对于反应原料100质量份为0~2000质量份的范围使用。
[0122]
反应催化剂可列举:苄基三乙基氯化铵、苄基三乙基溴化铵、苄基三甲基氯化铵、四甲基氯化铵、四甲基溴化铵、四甲基碘化铵、四甲基氢氧化铵、四乙基溴化铵、四丁基氯化铵、四丁基溴化铵、四丁基碘化铵、四丁基硫酸氢铵、三辛基甲基氯化铵、三丁基苄基氯化铵、三甲基苄基氯化铵、三甲基苄基氢氧化铵、n-月桂基氯化吡啶、n-月桂基氯化4-甲吡啶、n-月桂基氯化甲吡啶、三甲基苯基溴化铵、n-苄基氯化甲吡啶等季铵盐类;四丁基氯化鏻、四丁基溴化鏻、四苯基氯化鏻等季鏻盐;三[2-(2-甲氧基乙氧基)乙基]胺、三(3,6-二氧杂庚基)胺、三(3,6-二氧杂辛基)胺等叔胺类。使用量相对于原料为0.001~100重量%,宜为0.005~50重量%的范围。
[0123]
反应温度宜为-50℃至溶剂的沸点左右,为室温至150℃更佳。反应时间从0.1~100小时适当选择。
[0124]
就反应方法而言,有如下方法:将环氧化合物、异氰尿酸衍生物、催化剂一次性地加入;使环氧化合物与异氰尿酸衍生物分散或溶解后,将催化剂一次性地添加或以溶剂稀释并滴加;或使催化剂分散或溶解后,将环氧化合物与异氰尿酸衍生物一次性地添加或以溶剂稀释并滴加。反应结束后,可不进行精制等而直接使用,也可为了去除存在于体系内的未反应的原料、催化剂等而以有机溶剂稀释后,进行分液洗净并回收。
[0125]
此时所使用的有机溶剂,只要是可溶解反应产物且与水混合则会分离成2层者,则无特别限制,可列举:己烷、庚烷、苯、甲苯、二甲苯等烃类;乙酸乙酯、乙酸正丁酯、丙二醇甲醚乙酸酯等酯类;甲乙酮、甲基戊基酮、环己酮、甲基异丁基酮等酮类;二乙醚、二异丙醚、甲基-叔丁醚、乙基环戊基甲醚等醚类;二氯甲烷、氯仿、二氯乙烷、三氯乙烯等氯系溶剂类;及它们的混合物等。此时使用的洗净水,使用通常被称为去离子水、超纯水者即可。洗净次数为1次以上即可,即使洗净10次以上,也不一定会获得相应的洗净效果,故宜为约1~5次。
[0126]
分液洗净时为了去除未反应的异氰尿酸或酸性成分,也可利用碱性水溶液进行洗净。碱可列举:碱金属的氢氧化物、碱金属的碳酸盐、碱土金属的氢氧化物、碱土金属的碳酸盐、氨、及有机铵等。
[0127]
另外,为了去除分液洗净时体系内的金属杂质或碱成分,也可利用酸性水溶液进行洗净。酸可列举:盐酸、氢溴酸、硫酸、硝酸、磷酸、杂多酸等无机酸类;草酸、三氟乙酸、甲磺酸、苯磺酸、对甲苯磺酸、三氟甲磺酸等有机酸类等。
[0128]
利用碱性水溶液、酸性水溶液所为的分液水洗可仅进行其中一者,也可组合进行,考量去除金属杂质的观点,分液水洗宜以碱性水溶液、酸性水溶液的顺序进行。
[0129]
上述利用碱性水溶液、酸性水溶液所为的分液水洗后,也可接续利用中性水进行洗净。洗净次数只要进行1次以上即可,宜为约1~5次。中性水可使用上述去离子水、超纯水等。洗净次数为1次以上即可,但次数少的话,会有无法去除碱成分、酸性成分的情况。即使洗净10次以上,也不一定会获得相应的洗净效果,故宜为约1~5次。
[0130]
另外,分液操作后的反应产物,也可于减压或常压将溶剂进行浓缩干固或晶析操作,而以粉体形式回收,为了改善制备含硅的抗蚀剂下层膜形成用组成物时的操作性,也可
预先制成适度浓度的溶液状态。此时浓度只要不过浓,则粘度不会过高且不会损及操作性。只要不过稀,则溶剂的量不会过大而且是经济的。此时的浓度宜为0.1~50质量%,更佳为0.5~30重量%。
[0131]
此时的溶剂只要是可溶解获得的化合物者,则无特别限制,举具体例的话,可列举:环己酮、甲基-2-戊基酮等酮类;3-甲氧基丁醇、3-甲基-3-甲氧基丁醇、1-甲氧基-2-丙醇、1-乙氧基-2-丙醇等醇类;丙二醇单甲醚、乙二醇单甲醚、丙二醇单乙醚、乙二醇单乙醚、丙二醇二甲醚、二乙二醇二甲醚等醚类;丙二醇单甲醚乙酸酯、丙二醇单乙醚乙酸酯、乳酸乙酯、丙酮酸乙酯、乙酸丁酯、3-甲氧基丙酸甲酯、3-乙氧基丙酸乙酯、乙酸叔丁酯、丙酸叔丁酯、丙二醇单叔丁醚乙酸酯等酯类,它们可单独使用或将2种以上混合使用。
[0132]
step1-2或step2-2表示的反应可利用已知的方法轻易地进行,酰化剂宜为与乙酸、丙烯酸、甲基丙烯酸、苯甲酸、1-萘甲酸、2-萘甲酸、2-蒽甲酸、9-蒽甲酸等对应的酰氯或酸酐较佳。使用酰氯时,可于无溶剂或二氯甲烷、乙腈、甲苯、己烷等溶剂中,依序或同时加入step1-1或step2-1中获得的化合物与酰氯与三乙胺、吡啶、4-二甲基氨基吡啶等碱,视需要进行冷却或加热等而实施。又,使用酸酐时,可于甲苯等溶剂中,依序或同时加入step1-1或step2-1中获得的化合物与三乙胺、吡啶、4-二甲基氨基吡啶等碱,视需要进行冷却或加热等而实施。获得的反应产物可施以水洗等精制操作,并以粉体或溶液的形式回收。以溶液的形式回收时的溶剂,可例示就将step1-1、step2-1中获得的化合物以溶液的形式回收时所使用的溶剂所列举者,此时的浓度宜为0.1~50质量%,更佳为0.5~30重量%。
[0133]
此外,本发明中使用的通式(a-1)表示的化合物可单独使用1种或将2种以上组合使用。这些化合物的添加量,相对于基础聚合物(利用后述方法获得的热交联性聚硅氧烷)100质量份,宜为0.01~40质量份,更佳为0.1~30质量份,尤佳为1~20质量份。
[0134]
通过将如此的化合物使用于例如含硅的抗蚀剂下层膜,对于形成于其上的光致抗蚀剂图案展现良好的密接性,又,相对于形成于上部的抗蚀剂图案与形成于下部的例如有机膜的两者展现高蚀刻选择性,故将形成的光致抗蚀剂图案利用干蚀刻处理依序进行图案转印至含硅的抗蚀剂下层膜、有机下层膜时,能以良好的图案形状进行图案转印。借此,最终可将以上层抗蚀剂形成的图案以高精度转印至基板。
[0135]
[热交联性聚硅氧烷]
[0136]
本发明的含硅的抗蚀剂下层膜形成用组成物,含有上述通式(a-1)表示的化合物中的1种或2种以上,且含有热交联性聚硅氧烷(sx)。
[0137]
本发明中使用的热交联性聚硅氧烷(sx),宜含有下列通式(sx-1)表示的重复单元、下列通式(sx-2)表示的重复单元、及下列通式(sx-3)表示的部分结构中的任一者以上。
[0138]
[化23]
[0139][0140]
[化24]
[0141][0142]
[化25]
[0143][0144]
式中,r4、r5、r6可各自相同也可不同地为碳数1~30的1价有机基团。
[0145]
上述热交联性聚硅氧烷(sx),可通过将下列水解性单体(sm)进行水解缩合而制造。
[0146]
作为水解性单体(sm),具体而言,可例示:四甲氧基硅烷、四乙氧基硅烷、四丙氧基硅烷、四异丙氧基硅烷、三甲氧基硅烷、三乙氧基硅烷、三丙氧基硅烷、三异丙氧基硅烷、甲基三甲氧基硅烷、甲基三乙氧基硅烷、甲基三丙氧基硅烷、甲基三异丙氧基硅烷、乙基三甲氧基硅烷、乙基三乙氧基硅烷、乙基三丙氧基硅烷、乙基三异丙氧基硅烷、乙烯基三甲氧基硅烷、乙烯基三乙氧基硅烷、乙烯基三丙氧基硅烷、乙烯基三异丙氧基硅烷、丙基三甲氧基硅烷、丙基三乙氧基硅烷、丙基三丙氧基硅烷、丙基三异丙氧基硅烷、异丙基三甲氧基硅烷、异丙基三乙氧基硅烷、异丙基三丙氧基硅烷、异丙基三异丙氧基硅烷、丁基三甲氧基硅烷、丁基三乙氧基硅烷、丁基三丙氧基硅烷、丁基三异丙氧基硅烷、仲丁基三甲氧基硅烷、仲丁基三乙氧基硅烷、仲丁基三丙氧基硅烷、仲丁基三异丙氧基硅烷、叔丁基三甲氧基硅烷、叔丁基三乙氧基硅烷、叔丁基三丙氧基硅烷、叔丁基三异丙氧基硅烷、环丙基三甲氧基硅烷、环丙基三乙氧基硅烷、环丙基三丙氧基硅烷、环丙基三异丙氧基硅烷、环丁基三甲氧基硅烷、环丁基三乙氧基硅烷、环丁基三丙氧基硅烷、环丁基三异丙氧基硅烷、环戊基三甲氧基硅烷、环戊基三乙氧基硅烷、环戊基三丙氧基硅烷、环戊基三异丙氧基硅烷、环己基三甲氧基硅烷、环己基三乙氧基硅烷、环己基三丙氧基硅烷、环己基三异丙氧基硅烷、环己烯基三甲氧基硅烷、环己烯基三乙氧基硅烷、环己烯基三丙氧基硅烷、环己烯基三异丙氧基硅烷、环己烯基乙基三甲氧基硅烷、环己烯基乙基三乙氧基硅烷、环己烯基乙基三丙氧基硅烷、环己烯基乙基三异丙氧基硅烷、环辛基三甲氧基硅烷、环辛基三乙氧基硅烷、环辛基三丙氧基硅烷、环辛基三异丙氧基硅烷、环戊二烯基丙基三甲氧基硅烷、环戊二烯基丙基三乙氧基硅烷、环戊二烯基丙基三丙氧基硅烷、环戊二烯基丙基三异丙氧基硅烷、双环庚烯基三甲氧基硅烷、双环庚烯基三乙氧基硅烷、双环庚烯基三丙氧基硅烷、双环庚烯基三异丙氧基硅烷、双环庚基三甲氧基硅烷、双环庚基三乙氧基硅烷、双环庚基三丙氧基硅烷、双环庚基三异丙氧基硅烷、金刚烷基三甲氧基硅烷、金刚烷基三乙氧基硅烷、金刚烷基三丙氧基硅烷、金刚烷基三异丙氧基硅烷、苯基三甲氧基硅烷、苯基三乙氧基硅烷、苯基三丙氧基硅烷、苯基三异丙氧基硅烷、苄基三甲氧基硅烷、苄基三乙氧基硅烷、苄基三丙氧基硅烷、苄基三异丙氧基硅烷、甲氧基苄基三甲氧基硅烷、甲氧基苄基三乙氧基硅烷、甲氧基苄基三丙氧基硅烷、甲氧基苄基三异丙氧基硅烷、甲苯基三甲氧基硅烷、甲苯基三乙氧基硅烷、甲苯基三丙氧基
硅烷、甲苯基三异丙氧基硅烷、苯乙基三甲氧基硅烷、苯乙基三乙氧基硅烷、苯乙基三丙氧基硅烷、苯乙基三异丙氧基硅烷、萘基三甲氧基硅烷、萘基三乙氧基硅烷、萘基三丙氧基硅烷、萘基三异丙氧基硅烷、二甲基二甲氧基硅烷、二甲基二乙氧基硅烷、甲基乙基二甲氧基硅烷、甲基乙基二乙氧基硅烷、二甲基二丙氧基硅烷、二甲基二异丙氧基硅烷、二乙基二甲氧基硅烷、二乙基二乙氧基硅烷、二乙基二丙氧基硅烷、二乙基二异丙氧基硅烷、二丙基二甲氧基硅烷、二丙基二乙氧基硅烷、二丙基二丙氧基硅烷、二丙基二异丙氧基硅烷、二异丙基二甲氧基硅烷、二异丙基二乙氧基硅烷、二异丙基二丙氧基硅烷、二异丙基二异丙氧基硅烷、二丁基二甲氧基硅烷、二丁基二乙氧基硅烷、二丁基二丙氧基硅烷、二丁基二异丙氧基硅烷、二仲丁基二甲氧基硅烷、二仲丁基二乙氧基硅烷、二仲丁基二丙氧基硅烷、二仲丁基二异丙氧基硅烷、二叔丁基二甲氧基硅烷、二叔丁基二乙氧基硅烷、二叔丁基二丙氧基硅烷、二叔丁基二异丙氧基硅烷、二环丙基二甲氧基硅烷、二环丙基二乙氧基硅烷、二环丙基二丙氧基硅烷、二环丙基二异丙氧基硅烷、二环丁基二甲氧基硅烷、二环丁基二乙氧基硅烷、二环丁基二丙氧基硅烷、二环丁基二异丙氧基硅烷、二环戊基二甲氧基硅烷、二环戊基二乙氧基硅烷、二环戊基二丙氧基硅烷、二环戊基二异丙氧基硅烷、二环己基二甲氧基硅烷、二环己基二乙氧基硅烷、二环己基二丙氧基硅烷、二环己基二异丙氧基硅烷、二环己烯基二甲氧基硅烷、二环己烯基二乙氧基硅烷、二环己烯基二丙氧基硅烷、二环己烯基二异丙氧基硅烷、二环己烯基乙基二甲氧基硅烷、二环己烯基乙基二乙氧基硅烷、二环己烯基乙基二丙氧基硅烷、二环己烯基乙基二异丙氧基硅烷、二环辛基二甲氧基硅烷、二环辛基二乙氧基硅烷、二环辛基二丙氧基硅烷、二环辛基二异丙氧基硅烷、二环戊二烯基丙基二甲氧基硅烷、二环戊二烯基丙基二乙氧基硅烷、二环戊二烯基丙基二丙氧基硅烷、二环戊二烯基丙基二异丙氧基硅烷、双(双环庚烯基)二甲氧基硅烷、双(双环庚烯基)二乙氧基硅烷、双(双环庚烯基)二丙氧基硅烷、双(双环庚烯基)二异丙氧基硅烷、双(双环庚基)二甲氧基硅烷、双(双环庚基)二乙氧基硅烷、双(双环庚基)二丙氧基硅烷、双(双环庚基)二异丙氧基硅烷、二金刚烷基二甲氧基硅烷、二金刚烷基二乙氧基硅烷、二金刚烷基二丙氧基硅烷、二金刚烷基二异丙氧基硅烷、二苯基二甲氧基硅烷、二苯基二乙氧基硅烷、甲基苯基二甲氧基硅烷、甲基苯基二乙氧基硅烷、二苯基二丙氧基硅烷、二苯基二异丙氧基硅烷、三甲基甲氧基硅烷、三甲基乙氧基硅烷、二甲基乙基甲氧基硅烷、二甲基乙基乙氧基硅烷、二甲基苯基甲氧基硅烷、二甲基苯基乙氧基硅烷、二甲基苄基甲氧基硅烷、二甲基苄基乙氧基硅烷、二甲基苯乙基甲氧基硅烷、二甲基苯乙基乙氧基硅烷等。
[0147]
上述化合物较佳可例示:四甲氧基硅烷、四乙氧基硅烷、甲基三甲氧基硅烷、甲基三乙氧基硅烷、乙基三甲氧基硅烷、乙基三乙氧基硅烷、乙烯基三甲氧基硅烷、乙烯基三乙氧基硅烷、丙基三甲氧基硅烷、丙基三乙氧基硅烷、异丙基三甲氧基硅烷、异丙基三乙氧基硅烷、丁基三甲氧基硅烷、丁基三乙氧基硅烷、异丁基三甲氧基硅烷、异丁基三乙氧基硅烷、烯丙基三甲氧基硅烷、烯丙基三乙氧基硅烷、环戊基三甲氧基硅烷、环戊基三乙氧基硅烷、环己基三甲氧基硅烷、环己基三乙氧基硅烷、环己烯基三甲氧基硅烷、环己烯基三乙氧基硅烷、苯基三甲氧基硅烷、苯基三乙氧基硅烷、苄基三甲氧基硅烷、苄基三乙氧基硅烷、苯乙基三甲氧基硅烷、苯乙基三乙氧基硅烷、二甲基二甲氧基硅烷、二甲基二乙氧基硅烷、二乙基二甲氧基硅烷、二乙基二乙氧基硅烷、甲基乙基二甲氧基硅烷、甲基乙基二乙氧基硅烷、二丙基二甲氧基硅烷、二丁基二甲氧基硅烷、甲基苯基二甲氧基硅烷、甲基苯基二乙氧基硅
烷、三甲基甲氧基硅烷、二甲基乙基甲氧基硅烷、二甲基苯基甲氧基硅烷、二甲基苄基甲氧基硅烷、二甲基苯乙基甲氧基硅烷等。
[0148]
作为水解性单体(sm),与以上例示的化合物对应的上述r4、r5、r6表示的有机基团的另一例,可列举具有1个以上的碳-氧单键或碳-氧双键的有机基团。具体而言,为具有选自由醚键、酯键、烷氧基、羟基等构成的群组中的1个以上的基团的有机基团。其示例可列举下列通式(sm-r)表示者。
[0149]
(p-q
1-(s1)
v1-q
2-)
u-(t)
v2-q
3-(s2)
v3-q
4-[0150]
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
(sm-r)
[0151]
通式(sm-r)中,p为氢原子、环状醚基、羟基、碳数1~4的烷氧基、碳数1~6的烷基羰基氧基、或碳数1~6的烷基羰基,q1、q2、q3、及q4各自独立地为-c
qh(2q-p)
p
p-(式中,p与上述同样,p为0~3的整数,q为0~10的整数(但是,q=0表示为单键。)。),u为0~3的整数,s1与s2各自独立地表示-o-、-co-、-oco-、-coo-或-ocoo-。v1、v2、及v3各自独立地表示0或1。t为碳以外的2价原子、脂环、芳香环或杂环构成的2价基团。就t而言,也可含有氧原子等杂原子的脂环、芳香环或杂环的示例如下所示。t中与q2和与q3键结的位置并无特别限定,可考量立体性因素所致的反应性、反应所使用的市售试剂的取得性等而适当选择。
[0152]
[化26]
[0153]
[0154]
通式(sm-r)中的具有1个以上的碳-氧单键或碳-氧双键的有机基团的较佳例,可列举如下。此外,下式中,(si)是用以表示与si的键结位置而记载的。
[0155]
[化27]
[0156][0157]
[化28]
[0158][0159]
又,作为r4、r5、r6的有机基团的示例,也可使用含有硅-硅键的有机基团。具体而言可列举如下。
[0160]
[化29]
[0161][0162]
又,作为r4、r5、r6的有机基团的示例,也可使用具有会因酸分解的保护基的有机基团。具体而言,可列举日本特开2013-167669号公报的[0043]段落至[0048]段落所列举的有机基团、由日本特开2013-224279号公报的[0056]段落所示的硅化合物获得的有机基团。
[0163]
另外,作为r4、r5、r6的有机基团的示例,也可使用具有氟原子的有机基团。具体而言,可列举由日本特开2012-53253号公报的[0059]段落至[0065]段落所示的硅化合物获得的有机基团。
[0164]
就上述水解性单体(sm)而言,于上述部分结构中(si)表示的硅上键结有1个、2个或3个氯、溴、碘、乙酰氧基、甲氧基、乙氧基、丙氧基或丁氧基等作为水解性基团。
[0165]
[热交联性聚硅氧烷(sx)的合成方法]
[0166]
(合成方法1:酸催化剂)
[0167]
本发明中使用的热交联性聚硅氧烷(sx),可通过将1种水解性单体(sm)或2种以上的混合物,在酸催化剂的存在下进行水解缩合而制造。
[0168]
此时使用的酸催化剂可列举:甲酸、乙酸、草酸、马来酸、甲磺酸、苯磺酸、甲苯磺酸等有机酸;氢氟酸、盐酸、氢溴酸、硫酸、硝酸、过氯酸、磷酸等无机酸。催化剂的使用量相对于单体1摩尔,为1
×
10-6
~10摩尔,宜为1
×
10-5
~5摩尔,更佳为1
×
10-4
~1摩尔。
[0169]
由这些单体利用水解缩合获得热交联性聚硅氧烷(sx)时的水的量,就键结于单体的水解性取代基每1摩尔而言,宜添加0.01~100摩尔,更佳为0.05~50摩尔,又更佳为0.1~30摩尔。若为100摩尔以下,反应所使用的装置变小,是经济的。
[0170]
就操作方法而言,在催化剂水溶液中添加单体并使水解缩合反应开始。此时,可在催化剂水溶液中添加有机溶剂,也可将单体利用有机溶剂进行稀释,也可实施两种。反应温度为0~100℃,宜为5~80℃。宜为于单体滴加时将温度保持在5~80℃,之后在20~80℃使其熟成的方法较佳。
[0171]
可添加至催化剂水溶液中的有机溶剂、或可稀释单体的有机溶剂宜为:甲醇、乙醇、1-丙醇、2-丙醇、1-丁醇、2-丁醇、2-甲基-1-丙醇、丙酮、乙腈、四氢呋喃、甲苯、己烷、乙
酸乙酯、甲乙酮、甲基异丁基酮、环己酮、甲基戊基酮、丁二醇单甲醚、丙二醇单甲醚、乙二醇单甲醚、丁二醇单乙醚、丙二醇单乙醚、乙二醇单乙醚、丙二醇二甲醚、二乙二醇二甲醚、丙二醇单甲醚乙酸酯、丙二醇单乙醚乙酸酯、丙酮酸乙酯、乙酸丁酯、3-甲氧基丙酸甲酯、3-乙氧基丙酸乙酯、乙酸叔丁酯、丙酸叔丁酯、丙二醇单叔丁醚乙酸酯、γ-丁内酯及它们的混合物等。
[0172]
这些溶剂中,宜为水溶性者。例如可列举:甲醇、乙醇、1-丙醇、2-丙醇等醇类;乙二醇、丙二醇等多元醇;丁二醇单甲醚、丙二醇单甲醚、乙二醇单甲醚、丁二醇单乙醚、丙二醇单乙醚、乙二醇单乙醚、丁二醇单丙醚、丙二醇单丙醚、乙二醇单丙醚等多元醇缩合物衍生物;丙酮、乙腈、四氢呋喃等。其中尤其宜为沸点为100℃以下者。
[0173]
此外,有机溶剂的使用量相对于单体1摩尔,宜为0~1,000ml,尤其宜为0~500ml。有机溶剂的使用量少的话,反应容器变小,是经济的。
[0174]
之后,视需要进行催化剂的中和反应,得到反应混合物水溶液。此时,可使用于中和的碱性物质的量,相对于催化剂所使用的酸宜为0.1~2当量。该碱性物质只要是在水中呈现碱性者,则可为任意物质。
[0175]
然后,宜利用减压去除等从反应混合物去除水解缩合反应所生成的醇等副产物。此时将反应混合物进行加热的温度取决于所添加的有机溶剂与反应产生的醇等的种类,宜为0~100℃,更佳为10~90℃,又更佳为15~80℃。又,此时的减压度取决于欲去除的有机溶剂及醇等的种类、排气装置、冷凝浓缩装置及加热温度而有所不同,宜为大气压以下,更佳为以绝对压力计80kpa以下,又更佳为以绝对压力计50kpa以下。虽难以正确得知此时去除的醇量,但去除约80质量%以上所生成的醇等较为理想。
[0176]
然后,也可从反应混合物将水解缩合所使用的酸催化剂去除。作为去除酸催化剂的方法,将水与热交联性聚硅氧烷溶液混合,并利用有机溶剂萃取热交联性聚硅氧烷。此时所使用的有机溶剂,宜为可溶解热交联性聚硅氧烷,且与水混合则会分离成2层者。例如可列举:甲醇、乙醇、1-丙醇、2-丙醇、1-丁醇、2-丁醇、2-甲基-1-丙醇、丙酮、四氢呋喃、甲苯、己烷、乙酸乙酯、环己酮、甲基戊基酮、丁二醇单甲醚、丙二醇单甲醚、乙二醇单甲醚、丁二醇单乙醚、丙二醇单乙醚、乙二醇单乙醚、丁二醇单丙醚、丙二醇单丙醚、乙二醇单丙醚、丙二醇二甲醚、二乙二醇二甲醚、丙二醇单甲醚乙酸酯、丙二醇单乙醚乙酸酯、丙酮酸乙酯、乙酸丁酯、3-甲氧基丙酸甲酯、3-乙氧基丙酸乙酯、乙酸叔丁酯、丙酸叔丁酯、丙二醇单叔丁醚乙酸酯、γ-丁内酯、甲基异丁基酮、环戊基甲醚及它们的混合物等。
[0177]
另外,也可使用水溶性有机溶剂与水难溶性有机溶剂的混合物。例如宜为:甲醇-乙酸乙酯混合物、乙醇-乙酸乙酯混合物、1-丙醇-乙酸乙酯混合物、2-丙醇-乙酸乙酯混合物、丁二醇单甲醚-乙酸乙酯混合物、丙二醇单甲醚-乙酸乙酯混合物、乙二醇单甲醚-乙酸乙酯混合物、丁二醇单乙醚-乙酸乙酯混合物、丙二醇单乙醚-乙酸乙酯混合物、乙二醇单乙醚-乙酸乙酯混合物、丁二醇单丙醚-乙酸乙酯混合物、丙二醇单丙醚-乙酸乙酯混合物、乙二醇单丙醚-乙酸乙酯混合物、甲醇-甲基异丁基酮混合物、乙醇-甲基异丁基酮混合物、1-丙醇-甲基异丁基酮混合物、2-丙醇-甲基异丁基酮混合物、丙二醇单甲醚-甲基异丁基酮混合物、乙二醇单甲醚-甲基异丁基酮混合物、丙二醇单乙醚-甲基异丁基酮混合物、乙二醇单乙醚-甲基异丁基酮混合物、丙二醇单丙醚-甲基异丁基酮混合物、乙二醇单丙醚-甲基异丁基酮混合物、甲醇-环戊基甲醚混合物、乙醇-环戊基甲醚混合物、1-丙醇-环戊基甲醚混合
物、2-丙醇-环戊基甲醚混合物、丙二醇单甲醚-环戊基甲醚混合物、乙二醇单甲醚-环戊基甲醚混合物、丙二醇单乙醚-环戊基甲醚混合物、乙二醇单乙醚-环戊基甲醚混合物、丙二醇单丙醚-环戊基甲醚混合物、乙二醇单丙醚-环戊基甲醚混合物、甲醇-丙二醇甲醚乙酸酯混合物、乙醇-丙二醇甲醚乙酸酯混合物、1-丙醇-丙二醇甲醚乙酸酯混合物、2-丙醇-丙二醇甲醚乙酸酯混合物、丙二醇单甲醚-丙二醇甲醚乙酸酯混合物、乙二醇单甲醚-丙二醇甲醚乙酸酯混合物、丙二醇单乙醚-丙二醇甲醚乙酸酯混合物、乙二醇单乙醚-丙二醇甲醚乙酸酯混合物、丙二醇单丙醚-丙二醇甲醚乙酸酯混合物、乙二醇单丙醚-丙二醇甲醚乙酸酯混合物等,但组合不限于这些。
[0178]
此外,水溶性有机溶剂与水难溶性有机溶剂的混合比例可适当选择,相对于水难溶性有机溶剂100质量份,水溶性有机溶剂为0.1~1,000质量份,宜为1~500质量份,又更佳为2~100质量份。
[0179]
然后,也可利用中性水进行洗净。该水使用通常被称为去离子水、超纯水的水即可。该水的量相对于热交联性聚硅氧烷溶液1l,宜为0.01~100l,更佳为0.05~50l,又更佳为0.1~5l。就该洗净方法而言,将两种溶液放入相同容器并搅拌,然后静置将水层分离即可。洗净次数为1次以上即可,即使洗净10次以上,也不一定会获得相应的洗净效果,故宜为约1~5次。
[0180]
作为其他将酸催化剂去除的方法,可列举利用离子交换树脂的方法、以环氧乙烷、环氧丙烷等环氧化合物中和后予以去除的方法。这些方法可配合反应所使用的酸催化剂而适当选择。
[0181]
利用此时的水洗操作,热交联性聚硅氧烷的一部分会逃往水层,有时会获得实质上与分级操作同等的效果,故水洗次数、洗净水的量视催化剂去除效果与分级效果而适当选择即可。
[0182]
残留有酸催化剂的热交联性聚硅氧烷溶液及去除了酸催化剂的热交联性聚硅氧烷溶液,均可通过添加最终的溶剂并于减压下进行溶剂交换,来获得期望的热交联性聚硅氧烷溶液。此时溶剂交换的温度取决于欲去除的反应溶剂、萃取溶剂的种类,宜为0~100℃,更佳为10~90℃,又更佳为15~80℃。又,此时的减压度取决于欲去除的萃取溶剂的种类、排气装置、冷凝浓缩装置及加热温度而有所不同,宜为大气压以下,更佳为以绝对压力计80kpa以下,又更佳为以绝对压力计50kpa以下。
[0183]
此时,会有因溶剂变化而导致热交联性聚硅氧烷变得不稳定的情况。其取决于最终的溶剂与热交联性聚硅氧烷的相容性(compatibility)而会发生,为了防止此情况,也可加入日本特开2009-126940号公报[0181]~[0182]段落记载的具有环状醚作为取代基的1元或2元以上的醇作为稳定剂。添加量相对于溶剂交换前的溶液中的热交联性聚硅氧烷100质量份,为0~25质量份,宜为0~15质量份,更佳为0~5质量份,但有添加时宜为0.5质量份以上。溶剂交换前的溶液中若有必要,也可添加具有环状醚作为取代基的1元或2元以上的醇后再实施溶剂交换操作。
[0184]
热交联性聚硅氧烷浓缩到一定浓度以上的话,会有缩合反应进一步进行,变化成无法再溶解于有机溶剂的状态的风险,故宜预先制成适当浓度的溶液状态。又,过稀的话,溶剂的量变得过多,故预先制成适当浓度的溶液状态是经济且较佳的。此时的浓度宜为0.1~20质量%。
[0185]
添加至热交联性聚硅氧烷溶液中的最终的溶剂宜为醇系溶剂,尤其宜为乙二醇、二乙二醇、三乙二醇、丙二醇、二丙二醇、丁二醇等的单烷醚衍生物。具体而言,宜为丁二醇单甲醚、丙二醇单甲醚、乙二醇单甲醚、丁二醇单乙醚、丙二醇单乙醚、乙二醇单乙醚、丁二醇单丙醚、丙二醇单丙醚、乙二醇单丙醚、二丙酮醇等。
[0186]
这些溶剂若为主成分,则也可添加非醇系溶剂作为辅助溶剂。该辅助溶剂可例示:丙酮、四氢呋喃、甲苯、己烷、乙酸乙酯、环己酮、甲基戊基酮、丙二醇二甲醚、二乙二醇二甲醚、丙二醇单甲醚乙酸酯、丙二醇单乙醚乙酸酯、丙酮酸乙酯、乙酸丁酯、3-甲氧基丙酸甲酯、3-乙氧基丙酸乙酯、乙酸叔丁酯、丙酸叔丁酯、丙二醇单叔丁醚乙酸酯、γ-丁内酯、甲基异丁基酮、环戊基甲醚等。
[0187]
又,作为使用了酸催化剂的另一反应操作,于单体或单体的有机溶液中添加水或含水有机溶剂,使水解反应开始。此时催化剂可添加在单体或单体的有机溶液中,也可添加在水或含水有机溶剂中。反应温度为0~100℃,宜为10~80℃。宜为于水滴加时加热至10~50℃,之后升温至20~80℃使其熟成的方法较佳。
[0188]
使用有机溶剂时,宜为水溶性者,可列举:甲醇、乙醇、1-丙醇、2-丙醇、1-丁醇、2-丁醇、2-甲基-1-丙醇、丙酮、四氢呋喃、乙腈、丁二醇单甲醚、丙二醇单甲醚、乙二醇单甲醚、丁二醇单乙醚、丙二醇单乙醚、乙二醇单乙醚、丁二醇单丙醚、丙二醇单丙醚、乙二醇单丙醚、丙二醇二甲醚、二乙二醇二甲醚、丙二醇单甲醚乙酸酯、丙二醇单乙醚乙酸酯、丙二醇单丙醚等多元醇缩合物衍生物及它们的混合物等。
[0189]
有机溶剂的使用量相对于单体1摩尔,宜为0~1,000ml,尤其宜为0~500ml。有机溶剂的使用量少则反应容器变小,是经济的。获得的反应混合物的后处理可和前述方法同样地进行后处理,并获得热交联性聚硅氧烷。
[0190]
(合成方法2:碱催化剂)
[0191]
又,热交联性聚硅氧烷(sx),可通过将1种水解性单体(sm)或2种以上的混合物,在碱催化剂的存在下进行水解缩合而制造。
[0192]
此时使用的碱催化剂可列举:甲胺、乙胺、丙胺、丁胺、乙二胺、六亚甲基二胺、二甲胺、二乙胺、乙基甲胺、三甲胺、三乙胺、三丙胺、三丁胺、环己胺、二环己胺、单乙醇胺、二乙醇胺、二甲基单乙醇胺、单甲基二乙醇胺、三乙醇胺、二氮杂双环辛烷、二氮杂双环环壬烯、二氮杂双环十一烯、六亚甲基四胺、苯胺、n,n-二甲基苯胺、吡啶、n,n-二甲基氨基吡啶、吡咯、哌嗪、吡咯烷、哌啶、甲基吡啶、四甲基氢氧化铵、氢氧化胆碱、四丙基氢氧化铵、四丁基氢氧化铵、氨、氢氧化锂、氢氧化钠、氢氧化钾、氢氧化钡、氢氧化钙等。催化剂的使用量相对于硅单体1摩尔,为1
×
10-6
摩尔~10摩尔,宜为1
×
10-5
摩尔~5摩尔,更佳为1
×
10-4
摩尔~1摩尔。
[0193]
由上述单体利用水解缩合获得热交联性聚硅氧烷时的水的量,就键结于单体的水解性取代基每1摩尔而言,宜添加0.1~50摩尔。为50摩尔以下的话,反应所使用的装置变小,是经济的。
[0194]
就操作方法而言,在催化剂水溶液中添加单体并使水解缩合反应开始。此时,可在催化剂水溶液中添加有机溶剂,也可将单体利用有机溶剂进行稀释,也可实施两种。反应温度为0~100℃,宜为5~80℃。宜为于单体滴加时将温度保持在5~80℃,之后在20~80℃使其熟成的方法较佳。
[0195]
可添加至碱催化剂水溶液中的有机溶剂、或可稀释单体的有机溶剂,宜使用与就可添加至酸催化剂水溶液中者所例示的有机溶剂同样者。此外,为了经济地实施反应,有机溶剂的使用量相对于单体1摩尔宜为0~1,000ml。
[0196]
之后,视需要进行催化剂的中和反应,得到反应混合物水溶液。此时,可使用于中和的酸性物质的量,相对于催化剂所使用的碱性物质宜为0.1~2当量。该酸性物质只要是在水中呈现酸性者,则可为任意物质。
[0197]
然后,宜利用减压去除等从反应混合物去除水解缩合反应所生成的醇等副产物。此时将反应混合物进行加热的温度取决于所添加的有机溶剂与反应产生的醇等的种类,宜为0~100℃,更佳为10~90℃,又更佳为15~80℃。又,此时的减压度取决于欲去除的有机溶剂及醇的种类、排气装置、冷凝浓缩装置及加热温度而有所不同,宜为大气压以下,更佳为以绝对压力计80kpa以下,又更佳为以绝对压力计50kpa以下。虽难以正确得知此时去除的醇量,但去除约80质量%以上所生成的醇较为理想。
[0198]
然后,为了去除水解缩合所使用的碱催化剂,利用有机溶剂萃取热交联性聚硅氧烷。此时所使用的有机溶剂,宜为可溶解热交联性聚硅氧烷,且与水混合则会分离成2层者。例如可列举:甲醇、乙醇、1-丙醇、2-丙醇、1-丁醇、2-丁醇、2-甲基-1-丙醇、丙酮、四氢呋喃、甲苯、己烷、乙酸乙酯、环己酮、甲基戊基酮、丙二醇单甲醚、乙二醇单甲醚、丙二醇单乙醚、乙二醇单乙醚、丙二醇单丙醚、乙二醇单丙醚、丙二醇二甲醚、二乙二醇二甲醚、丙二醇单甲醚乙酸酯、丙二醇单乙醚乙酸酯、丙酮酸乙酯、乙酸丁酯、3-甲氧基丙酸甲酯、3-乙氧基丙酸乙酯、乙酸叔丁酯、丙酸叔丁酯、丙二醇单叔丁醚乙酸酯、γ-丁内酯、甲基异丁基酮、环戊基甲醚、及它们的混合物等。
[0199]
另外,也可使用水溶性有机溶剂与水难溶性有机溶剂的混合物。
[0200]
去除碱催化剂时使用的有机溶剂的具体例,可使用与就去除酸催化剂时所使用者具体例示的上述有机溶剂、水溶性有机溶剂与水难性有机溶剂的混合物同样者。
[0201]
此外,水溶性有机溶剂与水难溶性有机溶剂的混合比例可适当选择,相对于难溶性有机溶剂100质量份,水溶性有机溶剂为0.1~1,000质量份,宜为1~500质量份,又更佳为2~100质量份。
[0202]
然后,利用中性水洗净。该水使用通常被称为去离子水、超纯水的水即可。该水的量相对于热交联性聚硅氧烷溶液1l,为0.01~100l,宜为0.05~50l,更佳为0.1~5l。就该洗净方法而言,将两种溶液放入相同容器并搅拌,然后静置将水层分离即可。洗净次数为1次以上即可,即使洗净10次以上,也不一定会获得相应的洗净效果,故宜为约1~5次。
[0203]
通过在洗净完毕的热交联性聚硅氧烷溶液中,添加最终的溶剂,于减压下进行溶剂交换,而获得期望的热交联性聚硅氧烷溶液。此时溶剂交换的温度取决于欲去除的萃取溶剂的种类,宜为0~100℃,更佳为10~90℃,又更佳为15~80℃。又,此时的减压度取决于欲去除的萃取溶剂的种类、排气装置、冷凝浓缩装置及加热温度而有所不同,宜为大气压以下,更佳为以绝对压力计80kpa以下,又更佳为以绝对压力计50kpa以下。
[0204]
添加至热交联性聚硅氧烷溶液中的最终的溶剂宜为醇系溶剂,尤其宜为乙二醇、二乙二醇、三乙二醇等的单烷醚、丙二醇、二丙二醇等的单烷醚。具体而言,宜为丙二醇单甲醚、乙二醇单甲醚、丙二醇单乙醚、乙二醇单乙醚、丙二醇单丙醚、乙二醇单丙醚、二丙酮醇等。
[0205]
又,作为使用了碱催化剂的另一反应操作,于单体或单体的有机溶液中添加水或含水有机溶剂,使水解反应开始。此时催化剂可添加在单体或单体的有机溶液中,也可添加在水或含水有机溶剂中。反应温度为0~100℃,宜为10~80℃。宜为于水滴加时加热至10~50℃,之后升温至20~80℃使其熟成的方法较佳。
[0206]
可作为单体的有机溶液或含水有机溶剂使用的有机溶剂,宜为水溶性者,可列举:甲醇、乙醇、1-丙醇、2-丙醇、1-丁醇、2-丁醇、2-甲基-1-丙醇、丙酮、四氢呋喃、乙腈、丙二醇单甲醚、乙二醇单甲醚、丙二醇单乙醚、乙二醇单乙醚、丙二醇单丙醚、乙二醇单丙醚、丙二醇二甲醚、二乙二醇二甲醚、丙二醇单甲醚乙酸酯、丙二醇单乙醚乙酸酯、丙二醇单丙醚等多元醇缩合物衍生物及它们的混合物等。
[0207]
利用上述合成方法1或2获得的热交联性聚硅氧烷的分子量,不仅可通过单体的选择来调整,还可通过控制聚合时的反应条件来调整,宜使用重量平均分子量为100,000以下,更佳为200~50,000,又更佳为300~30,000者。重量平均分子量为100,000以下的话,不会有产生异物、发生涂布不均的情况。此外,关于上述重量平均分子量的数据,利用使用ri作为检测器,并使用四氢呋喃作为溶离溶剂的凝胶渗透层析法(gpc),并使用聚苯乙烯作为标准物质,以聚苯乙烯换算来表示分子量。
[0208]
本发明中使用的热交联性聚硅氧烷的物性,取决于水解缩合时使用的酸或碱催化剂的种类、反应条件而有所不同。因此,可配合作为目的的抗蚀剂下层膜的性能而适当选择。
[0209]
另外,可使用将1种或2种以上的水解性单体(sm)与下列通式(mm)表示的水解性金属化合物的混合物以利用了前述酸或碱催化剂的条件制得的聚硅氧烷衍生物作为抗蚀剂下层膜形成用组成物的成分。
[0210]
[化30]
[0211]
u(or7)
m7
(or8)
m8
ꢀꢀꢀꢀꢀꢀ
(mm)
[0212]
式中,r7、r8为碳数1~30的有机基团,m7 m8为依u的种类而决定的和价数相同的数,m7、m8为0以上的整数,u为周期表的iii族、iv族、或v族的元素,且排除碳及硅。
[0213]
作为上述通式(mm)表示的水解性金属化合物,可例示硼、铝、镓、钇、锗、钛、铪等的金属醇盐,具体而言可使用日本特开2020-118960号公报的[0107]~[0123]记载者。
[0214]
(酸产生剂)
[0215]
本发明的含硅的抗蚀剂下层膜形成用组成物中,可更掺合1种或2种以上的酸产生剂。酸产生剂只要是热酸产生剂、光酸产生剂、酸增殖剂等作为酸前驱体发挥作用的物质,则皆可使用,本发明中掺合的酸产生剂为是锍盐且会因高能量射线的作用而产生酸的光酸产生剂更佳。具体而言,可添加日本特开2007-199653号公报中的[0061]~[0085]段落记载的材料,但不限定于这些。
[0216]
上述酸产生剂可单独使用1种或将2种以上组合使用。添加酸产生剂时的添加量,相对于热交联性聚硅氧烷100份宜为0.05~50份,更佳为0.1~10份。
[0217]
[其他成分]
[0218]
(交联催化剂)
[0219]
本发明中,也可将交联催化剂(xc)掺合至含硅的抗蚀剂下层膜形成用组成物中。能掺合的交联催化剂,可列举下列通式(xc0)表示的化合物。
[0220]
lah
baꢀꢀ
(xc0)
[0221]
式中,l为锂、钠、钾、铷、铯、锍、錪、鏻或铵。a为非亲核性相对离子。a为1以上的整数,b为0或1以上的整数,且a b为非亲核性相对离子的价数。
[0222]
就作为具体的(xc0)的本发明中使用的交联催化剂而言,可列举下列通式(xc-1)的锍盐、下列通式(xc-2)的錪盐、下列通式(xc-3)的鏻盐、下列通式(xc-4)的铵盐、碱金属盐等、具有铵盐、锍盐、鏻盐、錪盐作为结构的一部分的聚硅氧烷(xc-10),具体而言,可添加日本特开2020-118960号公报中的[0124]~[0163]段落记载的材料等。
[0223]
[化31]
[0224][0225]
[化32]
[0226][0227]
式中,r
204
、r
205
、r
206
、r
207
各自表示碳数1~12的直链状、分支状或环状的烷基、烯基、氧代烷基或氧代烯基、碳数6~20的取代或非取代的芳基、或碳数7~12的芳烷基或芳基氧代烷基,这些基团的氢原子的一部分或全部也可被烷氧基取代等。又,r
205
与r
206
也可形成环,形成环时,r
205
、r
206
各自表示碳数1~6的亚烷基。a-表示非亲核性相对离子。r
208
、r
209
、r
210
、r
211
,与r
204
、r
205
、r
206
、r
207
同样,但也可为氢原子。r
208
与r
209
、r
208
与r
209
与r
210
也可形成环,形成环时,r
208
与r
209
及r
208
与r
209
与r
210
表示碳数3~10的亚烷基。
[0228]
上述交联催化剂(xc-1)、(xc-2)、(xc-3)、(xc-4)、(xc-10)可单独使用1种或将2种以上组合使用。交联催化剂的添加量,相对于基础聚合物(例如,利用上述方法获得的热交联性聚硅氧烷(sx))100质量份,宜为0.01~50质量份,更佳为0.1~40质量份。
[0229]
本发明的含硅的抗蚀剂下层膜形成用组成物中,也可更掺合下列原料。
[0230]
(有机酸)
[0231]
为了改善本发明的含硅的抗蚀剂下层膜形成用组成物的稳定性,宜添加碳数为1~30的1元或2元以上的有机酸。此时添加的酸可例示:甲酸、乙酸、丙酸、丁酸、戊酸、己酸、庚酸、辛酸、壬酸、癸酸、油酸、硬脂酸、亚麻油酸、次亚麻油酸、苯甲酸、邻苯二甲酸、间苯二甲酸、对苯二甲酸、水杨酸、三氟乙酸、单氯乙酸、二氯乙酸、三氯乙酸、草酸、丙二酸、甲基丙二酸、乙基丙二酸、丙基丙二酸、丁基丙二酸、二甲基丙二酸、二乙基丙二酸、琥珀酸、甲基琥珀酸、戊二酸、己二酸、伊康酸、马来酸、富马酸、柠康酸、柠檬酸等。尤其宜为草酸、马来酸、甲酸、乙酸、丙酸、柠檬酸等。又,为了保持稳定性,也可将2种以上的酸混合使用。
[0232]
有机酸的添加量,相对于本发明的含硅的抗蚀剂下层膜形成用组成物中含有的热交联性聚硅氧烷100质量份为0.001~25质量份,宜为0.01~15质量份,更佳为0.1~5质量
份。
[0233]
或也可掺合上述有机酸以使换算成本发明的含硅的抗蚀剂下层膜形成用组成物的ph较佳成为0≤ph≤7,更佳成为0.3≤ph≤6.5,又更佳成为0.5≤ph≤6。
[0234]
(水)
[0235]
本发明中,也可于组成物中添加水。添加水的话,本发明的含硅的抗蚀剂下层膜形成用组成物中的聚硅氧烷化合物进行水合,故光刻性能改善。本发明的含硅的抗蚀剂下层膜形成用组成物的溶剂成分中的水的含有率为超过0质量%且未达50质量%,尤其宜为0.3~30质量%,又更佳为0.5~20质量%。水的添加量为上述范围内的话,含硅的抗蚀剂下层膜的均匀性良好,不会有产生眼孔(eye hole),或光刻性能降低的风险。
[0236]
包含水的全部溶剂的使用量,相对于是基础聚合物的热交联性聚硅氧烷(sx)100质量份,宜为100~100,000质量份,尤其宜为200~50,000质量份。
[0237]
(稳定剂)
[0238]
另外,本发明中,可于组成物中添加稳定剂。稳定剂可添加具有环状醚作为取代基的1元或2元以上的醇。尤其添加日本特开2009-126940号公报[0181]~[0182]段落记载的稳定剂的话,可改善含硅的抗蚀剂下层膜形成用组成物的稳定性。
[0239]
(表面活性剂)
[0240]
另外,本发明中,可视需要于组成物中掺合表面活性剂。如此的表面活性剂,具体而言,可添加日本特开2009-126940号公报[0185]段落记载的材料。
[0241]
(高沸点溶剂)
[0242]
另外,本发明中,也可视需要于组成物中添加沸点为180度以上的高沸点溶剂。该高沸点溶剂可例示:1-辛醇、2-乙基己醇、1-壬醇、1-癸醇、1-十一醇、乙二醇、1,2-丙二醇、1,3-丁二醇、2,4-戊二醇、2-甲基-2,4-戊二醇、2,5-己二醇、2,4-庚二醇、2-乙基-1,3-己二醇、二乙二醇、二丙二醇、三乙二醇、三丙二醇、甘油、γ-丁内酯、三丙二醇单甲醚、二丙酮醇、乙酸正壬酯、乙酸乙二醇单乙醚、1,2-二乙酰氧基乙烷、1-乙酰氧基-2-甲氧基乙烷、1,2-二乙酰氧基丙烷、乙酸二乙二醇单甲醚、乙酸二乙二醇单乙醚、乙酸二乙二醇单-正丁醚、乙酸丙二醇单甲醚、乙酸丙二醇单丙醚、乙酸丙二醇单丁醚、乙酸二丙二醇单甲醚、乙酸二丙二醇单乙醚等。
[0243]
[图案形成方法]
[0244]
(图案形成方法1)
[0245]
本发明的图案形成方法之一是包含下列步骤的图案形成方法(所谓“多层抗蚀剂法”):于被加工体上使用涂布型有机膜材料来形成有机膜;于前述有机膜上使用本发明的含硅的抗蚀剂下层膜形成用组成物来形成含硅的抗蚀剂下层膜;于上述含硅的抗蚀剂下层膜上使用由光致抗蚀剂组成物构成的抗蚀剂上层膜用组成物来形成抗蚀剂上层膜;于上述抗蚀剂上层膜形成电路图案;将形成有上述电路图案的上述抗蚀剂上层膜作为掩膜,利用蚀刻将图案转印至上述含硅的抗蚀剂下层膜;将转印有上述图案的上述含硅的抗蚀剂下层膜作为掩膜,利用蚀刻对上述有机膜进行图案转印;及将转印有上述图案的上述有机膜作为掩膜,利用蚀刻将图案转印至上述被加工体。
[0246]
(图案形成方法2)
[0247]
又,本发明的图案形成方法是包含下列步骤的图案形成方法(所谓“多层抗蚀剂
法”):于被加工体上利用cvd法形成以碳作为主成分的有机硬掩膜;于前述硬掩膜上使用本发明的含硅的抗蚀剂下层膜形成用组成物来形成含硅的抗蚀剂下层膜;于上述含硅的抗蚀剂下层膜上使用由光致抗蚀剂组成物构成的抗蚀剂上层膜用组成物来形成抗蚀剂上层膜;于上述抗蚀剂上层膜形成电路图案;将形成有上述电路图案的上述抗蚀剂上层膜作为掩膜,利用蚀刻将图案转印至上述含硅的抗蚀剂下层膜;将转印有上述图案的上述含硅的抗蚀剂下层膜作为掩膜,利用蚀刻对上述硬掩膜进行图案转印;及将转印有上述图案的上述硬掩膜作为掩膜,利用蚀刻将图案转印至上述被加工体。
[0248]
于被加工体上(含硅的抗蚀剂下层膜之下)形成涂布型有机膜时,涂布型有机膜材料宜使用含有含芳香环的化合物者。使用如此的材料作为涂布型有机膜材料的话,可进一步抑制图案崩塌的发生。
[0249]
另一方面,于被加工体上(含硅的抗蚀剂下层膜之下)形成cvd硬掩膜时,可利用cvd法形成以碳作为主成分的硬掩膜,也可利用已知的方法进行。
[0250]
使用本发明的含硅的抗蚀剂下层膜形成用组成物形成抗蚀剂图案的话,如上述,通过使硬掩膜、有机膜的组合最适化,可抑制图案崩塌且以良好的形状在基板上形成以光致抗蚀剂形成的超微细图案。又,图案形成后所残存的含硅的抗蚀剂下层膜容易利用蚀刻等去除,故可抑制成为缺陷的原因的残渣,又,也可防止因过剩的蚀刻条件所致的基板损伤。
[0251]
形成含硅的抗蚀剂下层膜时,可通过利用旋涂法等将本发明的含硅的抗蚀剂下层膜形成用组成物涂布于被加工体上来进行。旋涂后,为了使溶剂蒸发,防止与由光致抗蚀剂组成物构成的抗蚀剂上层膜、由涂布型有机膜材料构成的有机膜的混合,并为了促进交联反应,而进行烘烤(热处理)。烘烤宜于50℃以上500℃以下、10~600秒的范围内进行,更佳为于100℃以上400℃以下、10~300秒的范围内进行。
[0252]
又,含硅的抗蚀剂下层膜的形成,也可通过利用上述同样的旋涂法等将本发明的含硅的抗蚀剂下层膜形成用组成物涂覆在被加工基板上,于氧浓度0.1%以上21%以下的环境中进行煅烧并使其硬化,而形成含硅的抗蚀剂下层膜。
[0253]
通过将本发明的含硅的抗蚀剂下层膜在如此的氧环境中进行煅烧,可获得充分硬化的膜。烘烤中的环境可为空气中,但为了使氧减少,预先封入n2、ar、he等钝性气体,可防止为有机膜的抗蚀剂下层膜氧化。为了防止氧化,须控制氧浓度,宜为1000ppm以下,更佳为100ppm以下。
[0254]
使用本发明的组成物所形成的含硅的抗蚀剂下层膜的厚度可适当选择,宜为1~300nm,更佳为1~200nm,尤佳为1~100nm。
[0255]
本发明的图案形成方法中,抗蚀剂上层膜用组成物只要是由化学增幅型光致抗蚀剂组成物构成者,则无特别限定。此外,本发明中,使用了碱显影液的正显影、使用了有机溶剂的显影液的负显影均可采用,故配合显影方法适当选择正型抗蚀剂上层膜材料、负型抗蚀剂上层膜材料即可。
[0256]
此外,正型图案形成方法中,在抗蚀剂上层膜形成、加热处理后,进行曝光,通常使用碱显影液来进行碱显影,获得正型抗蚀剂图案。又,曝光后宜进行曝光后烘烤(peb)。
[0257]
该碱显影液可使用四甲基氢氧化铵(tmah)水溶液等。
[0258]
又,负型图案形成方法中,在抗蚀剂上层膜形成、加热处理后,进行曝光,通常使用
有机溶剂来进行有机溶剂显影,获得负型抗蚀剂图案。又,曝光后宜进行peb。
[0259]
该有机溶剂的显影液,可使用含有选自4-甲基-2-戊醇、2-辛酮、2-壬酮、2-庚酮、3-庚酮、4-庚酮、2-己酮、3-己酮、二异丁基酮、甲基环己酮、苯乙酮、甲基苯乙酮、乙酸丙酯、乙酸丁酯、乙酸异丁酯、乙酸戊酯、乙酸丁烯酯、乙酸异戊酯、乙酸苯酯、甲酸丙酯、甲酸丁酯、甲酸异丁酯、甲酸戊酯、甲酸异戊酯、戊酸甲酯、戊烯酸甲酯、巴豆酸甲酯、巴豆酸乙酯、乳酸甲酯、乳酸乙酯、乳酸丙酯、乳酸丁酯、乳酸异丁酯、乳酸戊酯、乳酸异戊酯、2-羟基异丁酸甲酯、2-羟基异丁酸乙酯、苯甲酸甲酯、苯甲酸乙酯、乙酸苯酯、乙酸苄酯、苯基乙酸甲酯、甲酸苄酯、甲酸苯基乙酯、3-苯基丙酸甲酯、丙酸苄酯、苯基乙酸乙酯、乙酸-2-苯基乙酯中的1种以上作为成分的显影液等。
[0260]
本发明的图案形成方法,抗蚀剂上层膜的图案崩塌受到抑制,且可形成良好的图案形状。又,利用干蚀刻所为的朝含硅的抗蚀剂下层膜的图案转印优异,且图案化结束后所残存的含硅的抗蚀剂下层膜的去除容易,不易产生因残渣所致的缺陷,故尤其于微细图案的形成是实用的图案形成方法。
[0261]
于上述抗蚀剂上层膜形成电路图案的步骤中的图案形成,宜利用波长为10nm以上300nm以下的光学光刻、电子束所为的直接描绘、纳米压印或它们的组合来形成图案较佳。
[0262]
上述被加工体宜为半导体装置基板、金属膜、金属碳化膜、金属氧化膜、金属氮化膜、金属氧化碳化膜或金属氧化氮化膜。
[0263]
上述被加工体为如此者时,会更充分地发挥本发明的效果。
[0264]
构成上述被加工体的金属,宜为硅、镓、钛、钨、铪、锆、铬、锗、铜、银、金、铟、砷、钯、钽、铱、铝、铁、钼、钴或它们的合金。
[0265]
构成上述被加工体的金属为上述特定金属时,会更充分地发挥本发明的效果。
[0266]
[实施例]
[0267]
以下,举合成例及实施例与比较例具体地说明本发明,但本发明不受这些记载限定。此外,下列示例中%表示质量%,分子量mw是利用gpc测定获得的聚苯乙烯换算的重量平均分子量。
[0268]
[合成例]化合物(a1)~(a7)的合成
[0269]
化合物(a1)~(a7)的合成使用下列(b1)~(b5)作为原料。
[0270]
[化33]
[0271][0272]
(合成例1)
[0273]
化合物(a1)的合成
[0274]
[化34]
[0275][0276]
于氮气环境下,在安装有温度计、回流管的三口烧瓶中,加入(b1)20.0g、(b4)32.8g、bht(二丁基羟基甲苯)0.05g、pgme(1-甲氧基丙醇)150g。升温至内温80℃并制成均匀溶液后,加入苄基三乙基氯化铵1.00g,于110℃反应7小时。反应停止后,以mibk(甲基异丁基酮)400ml稀释后,移至分液漏斗,依序以1%氨水溶液100g洗净2次,以3%硝酸水溶液100g洗净2次,以超纯水100ml洗净5次。回收有机相并进行浓缩干固,借此得到化合物(a1)51.0g。
[0277]
(合成例2)
[0278]
化合物(a2)的合成
[0279]
[化35]
[0280][0281]
于氮气环境下,在安装有温度计、回流管的三口烧瓶中,加入(b2)20.0g、(b4)29.8g、bht0.05g、pgme150g。升温至内温80℃并制成均匀溶液后,加入苄基三乙基氯化铵1.00g(0.4mmol),于110℃反应7小时。反应停止后,以mibk400ml稀释后,移至分液漏斗,依序以1%氨水溶液100g洗净2次,以3%硝酸水溶液100g洗净2次,以超纯水100ml洗净5次。回收有机相并进行浓缩干固,借此得到化合物(a2)48.3g。
[0282]
(合成例3)
[0283]
化合物(a3)的合成
[0284]
[化36]
[0285][0286]
于氮气环境下,在安装有温度计、回流管的三口烧瓶中,加入(b2)20.0g、(b5)
29.2g、pgme150g。升温至内温80℃并制成均匀溶液后,加入苄基三乙基氯化铵1.0g,于110℃反应7小时。反应停止后,以mibk400ml稀释后,移至分液漏斗,依序以1%氨水溶液100g洗净2次,以3%硝酸水溶液100g洗净2次,以超纯水100ml洗净5次。回收有机相并进行浓缩干固,借此得到化合物(a3)46.7g。
[0287]
(合成例4)
[0288]
化合物(a4)的合成
[0289]
[化37]
[0290][0291]
于氮气环境下,在安装有温度计、回流管的三口烧瓶中,加入(b3)20.0g、(b4)42.2g、bht0.05g、pgme200g。升温至内温80℃并制成均匀溶液后,加入苄基三乙基氯化铵1.0g,于110℃反应7小时。反应停止后,以mibk500ml稀释后,移至分液漏斗,依序以1%氨水溶液125g洗净2次,以3%硝酸水溶液125g洗净2次,以超纯水125ml洗净5次。回收有机相并进行浓缩干固,借此得到化合物(a4)59.7g。
[0292]
(合成例5)
[0293]
化合物(a5)的合成
[0294]
[化38]
[0295][0296]
于氮气环境下,在安装有温度计、回流管的三口烧瓶中,加入(b3)20.0g、(b5)41.4g、pgme200g。升温至内温80℃并制成均匀溶液后,加入苄基三乙基氯化铵1.0g,于110℃反应7小时。反应停止后,以mibk500ml稀释后,移至分液漏斗,依序以1%氨水溶液125g洗
净2次,以3%硝酸水溶液125g洗净2次,以超纯水125ml洗净5次。回收有机相并进行浓缩干固,借此得到化合物(a5)60.2g。
[0297]
(合成例6)
[0298]
化合物(a6)的合成
[0299]
[化39]
[0300][0301]
加入化合物(a2)20.0g、吡啶11.3g、n-甲基吡咯烷酮80g,于氮气环境下、室温制成均匀溶液,缓慢滴加乙酸酐11.7g后,于40℃反应3小时。反应结束后,加入mibk200ml后,边于冰浴中冷却边缓慢加入超纯水100g,使反应淬灭。淬灭后,移至分液漏斗并去除水层,将有机层以2%nahco3水溶液100g洗净2次,以3%硝酸水溶液60g洗净2次,以纯水60g洗净6次后,将有机层进行减压干固。于残渣中加入thf60g,制成均匀溶液后,以己烷200g进行晶析。将沉降的结晶利用过滤分取,以己烷200g洗净2次并回收。将回收的结晶于40℃进行真空干燥,借此得到(a6)18.5g。
[0302]
(合成例7)
[0303]
化合物(a7)的合成
[0304]
[化40]
[0305][0306]
加入化合物(a5)20.0g、三乙胺11.1g、antage w-400 0.05g、n-甲基吡咯烷酮80g,于氮气环境下、冰浴中制成均匀溶液,缓慢滴加丙烯酰氯7.9g后,于室温反应3小时。反应结束后,加入mibk200ml后,边在冰浴中冷却边缓慢加入5%盐酸水溶液100g,使反应淬灭。淬灭后,移至分液漏斗并去除水层,将有机层以3%硝酸水溶液60g、纯水60g洗净6次后,将有机层进行减压干固。于残渣中加入thf60g制成均匀溶液后,以己烷300g进行晶析。将沉降的结晶利用过滤分取,以己烷200g洗净2次并回收。将回收的结晶于40℃进行真空干燥,借此
得到(a7)20.5g。
[0307]
[合成例]热交联性聚硅氧烷(c1)~(c3)的合成
[0308]
(合成例8)
[0309]
聚硅氧烷(c1)的合成
[0310]
[化41]
[0311][0312]
加入甲醇120g、10%硝酸0.1g及超纯水60g,于氮气环境下、40℃制成均匀溶液后,缓慢滴加四甲氧基硅烷49.4g、甲基三甲氧基硅烷20.4g及苯基三甲氧基硅烷5.0g的混合物。滴加后,于40℃进行12小时的水解缩合反应。反应结束后,加入pgee(丙二醇乙醚)600g,将水分及副产物醇馏去,以聚硅氧烷化合物(c1)的pgee溶液400g(化合物浓度10%)的形式回收。测定聚硅氧烷化合物(c1)的聚苯乙烯换算分子量,结果mw=2600。
[0313]
(合成例9)
[0314]
聚硅氧烷(c2)的合成
[0315]
[化42]
[0316][0317]
加入甲醇120g、10%硝酸0.1g及超纯水60g,于氮气环境下、40℃制成均匀溶液后,缓慢滴加四甲氧基硅烷38.1g、甲基三甲氧基硅烷30.6g及3-环氧丙氧基丙基三甲氧基硅烷5.9g的混合物。滴加后,于40℃进行12小时的水解缩合反应。反应结束后,加入pgee(丙二醇乙醚)600g,将水分及副产物醇馏去,以聚硅氧烷化合物(c2)的pgee溶液440g(化合物浓度10%)的形式回收。测定聚硅氧烷化合物(c2)的聚苯乙烯换算分子量,结果mw=2900。
[0318]
(合成例10)
[0319]
聚硅氧烷(c3)的合成
[0320]
[化43]
[0321][0322]
加入乙醇1400g、超纯水700g及25%四甲基氢氧化铵50g,于氮气环境下、40℃制成均匀溶液。缓慢滴加2-(3,4-环氧环己基)乙基三甲氧基硅烷138.6g与苯基三甲氧基硅烷37.2g的混合物后,于40℃反应2小时。反应结束后,加入乙酸35g使反应停止,并于减压将乙醇馏去。于馏去后的溶液中加入乙酸乙酯2000ml并分取水层,将有机层以超纯水400ml洗净2次后,加入pgmea(丙二醇单甲醚乙酸酯)1000g,将水分及低沸点溶剂馏去,借此以聚硅氧烷化合物(c3)的pgmea溶液600g(化合物浓度20%)的形式回收。测定聚硅氧烷化合物(c3)的聚苯乙烯换算分子量,结果mw=2800。
[0323]
[实施例、比较例]
[0324]
将上述合成例中获得的化合物(a1)~(a7)、聚硅氧烷化合物(c1)~(c3)、交联催化剂、光酸产生剂(表3记载的pag1~3)、酸(马来酸)、溶剂、水,以表1~3所示的比例混合,利用0.1μm的氟树脂制的过滤器进行过滤,借此分别制备含硅的抗蚀剂下层膜形成用组成物溶液,并分别定义为sol.1~sol.41。
[0325]
[表1]
[0326][0327]
[表2]
[0328][0329]
[表3]
[0330][0331]
所使用的交联催化剂如下。
[0332]
tpsno3…
硝酸三苯基锍
[0333]
qbano3…
硝酸四丁基铵
[0334]
teahnf
…
全氟丁磺酸三乙基铵
[0335]
所使用的溶剂如下。
[0336]
pgee
…
丙二醇乙醚
[0337]
pgmea
…
丙二醇甲醚乙酸酯
[0338]
所使用的光酸产生剂如下列表4所示。
[0339]
[表4]
[0340][0341]
[实施例1-1~1-36、比较例1-1~1-5]
[0342]
(euv图案化试验)
[0343]
在硅晶圆(si基板)上旋涂含硅的抗蚀剂下层膜形成用组成物sol.1~41,于220℃加热60秒,制作膜厚25nm的含硅的抗蚀剂下层膜film1~41(聚硅氧烷抗蚀剂下层膜)。
[0344]
然后,将以表5的比例溶解有下列成分的抗蚀剂材料旋涂在film1~41上,使用加热板于105℃预烘60秒,制作膜厚35nm的抗蚀剂膜。将其使用asml公司制euv扫描曝光机nxe3300(na0.33、σ0.9/0.6、四极照明、晶圆上尺寸为节距36nm的l/s图案)进行曝光,在加热板上于100℃进行60秒peb,以2.38质量%tmah水溶液显影30秒,得到尺寸18nm的线。
[0345]
使用日立先端科技(股)制的测长sem(cg5000)测定该线尺寸,于线宽15nm未观察到图案崩塌时评价为良好,观察到图案崩塌时评价为不良。又,利用日立先端科技(股)制电子显微镜(s-4800)观测剖面形状(图案形状),未观察到拖尾形状时评价为良好,观察到明显的拖尾形状时评价为不良。关于图案粗糙度,利用日立先端科技(股)制电子显微镜(cg4000)进行观测。
[0346]
抗蚀剂材料所使用的聚合物、淬灭剂、增感剂、表面活性剂及有机溶剂如下。
[0347]
[化44]
[0348][0349]
[化45]
[0350][0351]
表面活性剂:3m公司制fc-4430
[0352]
pgmea:丙二醇单甲醚乙酸酯
[0353]
cyho:环己酮
[0354]
pgme:丙二醇单甲醚
[0355]
[表5]
[0356][0357]
上述试验的结果示于表6~7。
[0358]
[表6]
[0359][0360]
[表7]
[0361][0362]
如表6~7所示,确认到添加了本发明的含硅的抗蚀剂下层膜形成用组成物所必需的化合物(a1)~(a7)的实施例1-1~1-36中,观察到抑制图案崩塌的效果及良好的图案形状,且图案粗糙度也良好。
[0363]
另一方面,比较例1-1~1-3中,由于不含本发明中使用的通式(a-1)表示的化合物,故发生了图案崩塌,又,图案形状及图案粗糙度差。另外,比较例1-4、1-5中,通过添加pag,观察到图案形状的改善,但由于未添加通式(a-1)表示的化合物,故观察到图案崩塌及图案粗糙度的劣化。由此可知,通过添加本发明中使用的化合物(a1)~(a7),可改善线宽15nm以上的微细图案的崩塌,不仅如此,于改善图案的形状及改善图案粗糙度也是有效的。
[0364]
此外,本发明不限定于上述实施形态。上述实施形态是例示的,具有与本发明的权利要求记载的技术思想实质相同的构成,且发挥同样的作用效果者,均包含在本发明的技术范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。