生产环状胍衍生物的方法
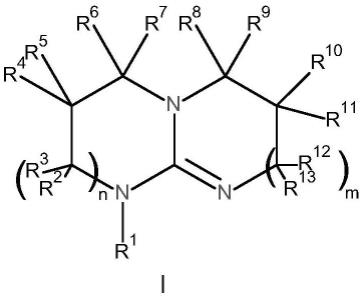
1.本发明涉及一种通过使三胺在c1源和固体材料存在下在气相或液相中在惰性气氛下反应而生产式i的环状胍衍生物或其混合物的方法:
[0002][0003]
其中r
1-r
13
独立地选自h和c
1-c4烷基的式i的环状胍衍生物是有价值的活性化学产品。它们用作许多不同化学反应的催化剂。用于合成双环胍的公开方法通常是复杂的,通常涉及使用多步合成工艺和/或要求使用太过昂贵的原料,后者可能如us-a1 2009/0281314中所提到的那样以各种方式有害。
[0004]
此外,环状胍衍生物,如7-甲基-1,5,7-三氮杂双环[5.5.0]癸-5-烯(me-tbd)—用有机酸处理而得到阳离子性1,5,7-三氮杂双环[4.4.0]癸-5-烯结构部分和阴离子—可以用作离子液体。这些离子液体如wo 2018/138416所述可以在制备纤维素纤维或薄膜的方法中用作溶剂。它们显示出良好的水热稳定性并且能够溶解木浆。所得溶液用于纺丝织物纤维。
[0005]
wo 2019/030197描述了一种通过使三胺与co2反应而产生环状脲单元的方法。没有公开在固体材料或有机强碱存在下在气相或液相中使用三胺与co2产生环状胍衍生物。
[0006]
us-a1 2009/0281314描述了一种通过将包含尿素和脱水剂如六甲基二硅氮烷(hmds)、四乙氧基硅烷(teos)、二硅氮烷、烷氧基取代硅烷或其组合的反应混合物加热至≥90℃的温度而生产双环胍的方法。us-a1 2009/0281314没有公开在气相或液相中在固体材料存在下生产双环胍的方法。us-a1 2009/0281314也没有公开在仅仅一个工艺步骤中生产环状胍衍生物的方法。
[0007]
因此,本发明的目的是要提供一种生产式i的环状胍衍生物的方法,该方法环境友好,比上述方法产生更少废物并且最多具有3个工艺步骤,优选一个工艺步骤。
[0008]
出人意料地发现这由一种通过在至少90℃的温度和1-320巴的压力下使三胺在c1源和固体材料存在下在气相或液相中在惰性气氛下反应而生产式i的环状胍衍生物或其混合物的方法实现:
[0009][0010]
其中r
1-r
13
独立地选自h和c
1-c4烷基,以及
[0011]
n和m是独立地选自0和1的自然数。
[0012]
当该固体材料选自惰性材料和固体酸催化剂时,本发明方法是有利的。
[0013]
当该固体材料是选自硅酸铝、氧化铝和沸石的固体酸催化剂时,本发明方法是有利的。
[0014]
当将该三胺和该c1源溶于选自甲醇、乙醇、异丙醇、丁醇、二甘醇、丁基二甘醇、四氢呋喃、二甘醇二甲醚、丙二醇二甲醚(proglyme)、三甘醇二甲醚、四甘醇二甲醚、甲苯、二氯苯、n,n-二甲基甲酰胺和n-甲基吡咯烷酮的溶剂中时,本发明方法是有利的。
[0015]
当该溶剂是甲醇、二甘醇或丁基二甘醇时,本发明方法是有利的。
[0016]
当该三胺选自n-(3-氨基丙基)-1,3-丙二胺(dpta)、n-(2-氨基乙基)-1,2-乙二胺(deta)和n-(2-氨基乙基)-1,3-丙二胺时,本发明方法是有利的。
[0017]
当该c1源选自碳酸二甲酯、尿素、二甲基甲酰胺二甲缩醛、二氧化碳、碳酸亚乙酯、碳酸亚丙酯、光气和氨基氰时,本发明方法是有利的。
[0018]
当该方法提供下面三个步骤时,本发明方法是有利的:
[0019]
i)使该三胺在c1源存在下在至少90℃的温度下反应成式ii的脲化合物:
[0020][0021]
其中r
1-r
13
、n和m具有式i中的相同含义,
[0022]
ii)将步骤i)的式ii的粗脲化合物溶于溶剂中,以及
[0023]
iii)在气相或液相中在惰性气氛下在固体材料存在下在至少120℃的温度
[0024]
和1-320巴范围内的压力下环化来自步骤ii)的式ii的粗脲化合物。
[0025]
当该方法在气相中在至少150℃的温度和1-35巴的压力下进行时,本发明方法是有利的。
[0026]
当在本发明方法的步骤i中存在催化量的有机碱或酸时,本发明方法是有利的。
[0027]
当本发明方法的步骤ii)的溶剂选自甲醇、乙醇、异丙醇、丁醇、二甘醇、丁基二甘醇、四氢呋喃、二甘醇二甲醚、丙二醇二甲醚、三甘醇二甲醚、四甘醇二甲醚、甲苯、二氯苯、n,n-二甲基甲酰胺和n-甲基吡咯烷酮时,本发明方法是有利的。
[0028]
当将选自甲醇、乙醇、异丙醇、丁醇、二甘醇、丁基二甘醇、四氢呋喃、二甘醇二甲醚、丙二醇二甲醚、三甘醇二甲醚、四甘醇二甲醚、甲苯、二氯苯、n,n-二甲基甲酰胺和n-甲基吡咯烷酮的溶剂用于本发明方法的步骤i中时,本发明方法是有利的。
[0029]
当若步骤ii)和步骤i)的溶剂相同的话,则本发明方法的步骤ii)包括在步骤i)中时,本发明方法是有利的。
[0030]
当式i的环状胍衍生物是1,5,7-三氮杂双环[4.4.0]癸-5-烯和7-甲基-1,5,7-三氮杂双环[4.4.0]癸-5-烯的混合物时,本发明方法是有利的。
[0031]
当式i的环状胍衍生物是1,2,3,5,6,7-六氢咪唑并[1,2-a]嘧啶和8-甲基-1,2,3,5,6,7-六氢咪唑并[1,2-a]嘧啶的混合物时,本发明方法是有利的。
[0032]
当式i的环状胍衍生物是2,3,5,6-四氢-1h-咪唑并[1,2-a]咪唑和1-甲基-2,3,5,6-四氢咪唑并[1,2-a]咪唑的混合物时,本发明方法是有利的。
[0033]
使用本发明方法生产式i的环状胍衍生物或其混合物:
[0034][0035]
在该式i中n和m为独立地选自0和1的自然数,优选m和n是相同的自然数,特别优选m和n为1。
[0036]
式i中的r
1-r
13
独立地选自h和c
1-c4烷基。c
1-c4烷基是指甲基、乙基、异丙基、正丙基、正丁基、异丁基和叔丁基。r
1-r
13
优选独立地选自h和甲基,特别优选r
2-r
13
是h且r1选自h和甲基。
[0037]
可以由本发明方法得到的式i的优选环状胍衍生物选自1,5,7-三氮杂双环[4.4.0]癸-5-烯、7-甲基-1,5,7-三氮杂双环[4.4.0]癸-5-烯、1,2,3,5,6,7-六氢咪唑并[1,2-a]嘧啶、8-甲基-1,2,3,5,6,7-六氢咪唑并[1,2-a]嘧啶、2,3,5,6-四氢-1h-咪唑并[1,2-a]咪唑和1-甲基-2,3,5,6-四氢咪唑并[1,2-a]咪唑。
[0038]
式i的环状胍衍生物的优选混合物是包含1,5,7-三氮杂双环[4.4.0]癸-5-烯和7-甲基-1,5,7-三氮杂双环[4.4.0]癸-5-烯、1,2,3,5,6,7-六氢咪唑并[1,2-a]嘧啶和8-甲基-1,2,3,5,6,7-六氢咪唑并[1,2-a]嘧啶或者2,3,5,6-四氢-1h-咪唑并[1,2-a]咪唑和1-甲基-2,3,5,6-四氢咪唑并[1,2-a]咪唑的那些。
[0039]
在本发明方法中,三胺与c1源反应。用于本发明方法的三胺选自n-(3-氨基丙基)-1,3-丙二胺(dpta)、n-(2-氨基乙基)-1,2-乙二胺(deta)和n-(2-氨基乙基)-1,3-丙二胺。优选使用n-(3-氨基丙基)-1,3-丙二胺(dpta)。
[0040]
用于本发明方法中的c1源选自碳酸二甲酯、尿素、二甲基甲酰胺二甲缩醛、二氧化碳、碳酸亚乙酯、碳酸亚丙酯、光气和氨基氰。优选使用碳酸二甲酯、尿素、二氧化碳、碳酸亚乙酯或碳酸亚丙酯。更优选使用碳酸二甲酯、尿素或二氧化碳。
[0041]
在本发明方法的过程中,c1源总是过量使用,这意味着c1源与所用三胺相比以1.01-1.50摩尔当量,优选1.01-1.20摩尔当量的量使用。当将二氧化碳用作c1源时,与所用三胺相比使用在10-500摩尔当量范围内的大过量二氧化碳。
[0042]
对本发明方法而言,起始产品三胺和c1源可以溶于溶剂中或者可以在没有溶剂下反应。优选三胺与c1源在没有溶剂下的反应。若使用溶剂,则该溶剂是选自甲醇、乙醇、异丙醇、丁醇、二甘醇和丁基二甘醇的质子性溶剂或者选自四氢呋喃、二甘醇二甲醚、丙二醇二甲醚、三甘醇二甲醚、四甘醇二甲醚、甲苯、二氯苯、n,n-二甲基甲酰胺和n-甲基吡咯烷酮的非质子溶剂。优选选自甲醇、丁基二甘醇和四氢呋喃的溶剂。特别优选诸如甲醇、二甘醇和丁基二甘醇的溶剂。
[0043]
本发明方法可以在气相或液相中用固体材料操作。优选在气相中用固体材料操作。
[0044]
将该固体材料置于反应器中。本发明方法原则上适合常用于在非均相催化剂上的反应—涉及将一种气态反应物和适用的话,一种或多种液态反应物供入反应器中—的耐压反应器。这些包括常用于气相和液相反应的常规反应器,例如管式反应器、管壳式反应器和气体循环反应器。管式反应器的具体实施方案是轴式反应器。该类反应器对本领域熟练技术人员而言原则上是已知的。更具体而言,使用具有垂直纵轴的圆筒形反应器,其在该反应器的底部或顶部具有用于供入包含至少一种气态组分和适用的话,至少一种液态组分的反应物混合物的输入装置或多个输入装置。需要的话,可以额外经由至少一个其他进料装置将气态和/或液态反应物的子流供入该反应器中。
[0045]
在优选的气相反应中,起始化合物如三胺、c1源、惰性气体和任选地,溶剂在该固体材料进入该反应器中之前在蒸发器中蒸发。该蒸发器可以是在该反应器内部的蒸发器床,其通过反应器加热而加热并且由选自拉西环、玻璃球、钢丝网或丝网环的惰性材料制成,或者它可以是在反应器顶部前方位于该反应器外部的额外油或蒸汽加热蒸发器装置。蒸发温度在80-280℃,优选120-250℃范围内。在蒸发过程中压力在1-35,优选1-25巴范围内。
[0046]
对于气相反应,起始化合物与固体材料在蒸发之后接触。对于液相反应,起始化合物如三胺、c1源、惰性气体和任选地,溶剂直接与固体材料接触。若在本发明方法的步骤ii中将式ii化合物溶于溶剂中,则这在供入固体材料之前在该反应器内部或外部发生。气相或液相通过该反应器的流动方向是垂直的。该流动方向可以从该反应器顶部到底部或者反之亦然。优选从该反应器顶部到底部的流动方向。
[0047]
在气相或液相中使用的固体材料选自惰性材料、固体酸催化剂或者惰性材料和固体酸催化剂的混合物或层。该惰性材料选自拉西环、玻璃球、钢丝网、丝网环和陶瓷材料。该固体酸催化剂选自硅酸铝、氧化铝、沸石或这些固体酸的混合物。固体材料可以具有不同的形状和尺寸。本领域熟练技术人员已知的该固体材料的所有形状和尺寸是可能的,若它们允许恒定流过该反应器的话。这些成型体选自片剂、挤出物、裂片、球形材料。与材料种类和尺寸无关,这些成型体的直径在1-20mm范围内。
[0048]
该固体材料固定在反应器内部并且为反应器床。该反应器床可以仅包括惰性材料或者包括惰性材料和一种或多种固体酸催化剂。优选的反应器床包括惰性材料和一种或多种固体酸催化剂。作为反应器床的一部分,惰性材料位于反应器床开始和结束处以将该床在反应器内部的位置固定。若使用固体酸催化剂,则该固体酸催化剂位于反应器床开始和结束处的惰性材料之间。该固体酸催化剂可以是单一固体酸催化剂或者两种或更多种混合或以分层排列的不同固体酸催化剂,其中惰性材料可以位于各层之间。优选的反应器床包括反应器床开始和结束处的惰性材料以及选自硅酸铝、氧化铝及其混合物的固体酸催化剂。
[0049]
在该气相和/或液相反应过程中,使用选自n2和氩气的惰性气体。可以用该惰性气体控制该反应的空速。空速通常在50-10000l/h范围内。
[0050]
该反应的温度为至少90℃,优选在150-350℃,更优选220-300℃范围内。在本发明方法过程中的压力在1-100巴范围内。在本发明方法的气相反应过程中,压力优选在1-35巴范围内。在本发明方法的液相反应过程中,优选的压力等于或小于100巴。
[0051]
本发明方法可以是一步、两步或三步方法。在一步方法中,该三胺、该c1源、该惰性气体和任选地,该溶剂在气相或液相中直接反应成式i化合物之一或式i化合物的混合物。在两步方法中,步骤i的溶剂与步骤ii的相同。在这种情况下步骤ii包括在步骤i中并且可以将式ii的脲化合物直接引入环化的步骤iii中。若在步骤i中不使用溶剂或者若在步骤ii中使用不同于步骤i的另一溶剂,则本发明方法以三步操作。优选本发明方法的该单步和三步方法。特别优选该单步方法。
[0052]
在本发明的另一实施方案中,该反应可以分成不止一步。优选三步。在第一步中使该三胺与该c1源反应成式ii的相应脲化合物:
[0053][0054]
其中r
1-r
13
、m和n具有式i中的相同含义。在三胺和c1源在本发明方法的步骤i中的反应中,可以使用催化量的有机碱或有机酸。催化量是在所用三胺的0.01-10mol%,优选0.1-5mol%范围内的量。有机碱或有机酸可以用于本发明方法的步骤i中。有机碱选自pka值为至少15的有机强碱。有机酸选自磺酸或羧酸。特别优选含氮有机碱,如胍类。
[0055]
该三胺和该c1源的反应可以在溶剂存在下或者在没有溶剂下进行。优选没有溶剂的反应。若使用溶剂,则它是质子性溶剂或非质子溶剂。用于步骤i的质子性溶剂选自甲醇、乙醇、异丙醇、丁醇、二甘醇和丁基二甘醇。用于步骤i的非质子溶剂选自四氢呋喃、二甘醇二甲醚、丙二醇二甲醚、三甘醇二甲醚、四甘醇二甲醚、甲苯、二氯苯、n,n-二甲基甲酰胺和n-甲基吡咯烷酮。该溶剂优选选自甲醇、二甘醇、丁基二甘醇和四氢呋喃。特别优选甲醇或丁基二甘醇作为溶剂。
[0056]
步骤i的反应的反应温度为至少90℃。若该c1源不是气体,则步骤i中的压力为大
气压力。若该c1源是co2,则在本发明方法的步骤i中压力在20-100巴范围内。
[0057]
在本发明方法的步骤i之后可以得到式ii的粗脲化合物。“粗”是指在该混合物中含有少量副产物。这些副产物选自起始产品,如三胺、c1源、烷基化的环状脲衍生物以及少量其中r1为h的式i化合物。少量是与在本发明方法结束时其中r1为h的式i化合物的量相比更小的那些量。
[0058]
少量副产物根据式ii的脲化合物在0.01-10重量%范围内。将所得式ii的粗脲化合物溶于溶剂中。该溶剂可以是质子性溶剂或非质子溶剂。它选自甲醇、乙醇、异丙醇、丁醇、二甘醇、丁基二甘醇、四氢呋喃、二甘醇二甲醚、丙二醇二甲醚、三甘醇二甲醚、四甘醇二甲醚、甲苯、二氯苯、n,n-二甲基甲酰胺和n-甲基吡咯烷酮。该溶剂优选选自甲醇、二甘醇、丁基二甘醇和四氢呋喃。特别优选的溶剂是甲醇、二甘醇和丁基二甘醇。若在步骤i中使用溶剂,则步骤ii的优选溶剂与步骤i的相同。
[0059]
然后将式ii的粗脲化合物与惰性气体和该溶剂一起供入填充有固体材料的反应器的液相或气相中以环化。若环化在气相中进行,则蒸发器位于反应器床之前。这可以在该反应器顶部或者在该反应器内部于反应器床之前进行。在该蒸发器中该粗脲和该溶剂一起蒸发,然后将该合并气体与惰性气体一起供入包括该固体材料的反应器床中。若环化在液相中进行,则首先将式ii的粗脲化合物溶于溶剂中,然后与惰性气体一起供入该反应器顶部或底部。
[0060]
在本发明方法的步骤iii中环化的温度为至少120℃,优选至少150℃,更优选在150-300℃范围内。在步骤iii过程中的压力在1-320巴,优选1-200巴范围内。
[0061]
优选的步骤iii在气相中在至少150℃的温度和1-35巴的压力下进行。
[0062]
通常得到本发明方法的式i的环状胍衍生物的混合物。优选的混合物包含1,5,7-三氮杂双环[4.4.0]癸-5-烯和7-甲基-1,5,7-三氮杂双环[4.4.0]癸-5-烯、1,2,3,5,6,7-六氢咪唑并[1,2-a]嘧啶和8-甲基-1,2,3,5,6,7-六氢咪唑并[1,2-a]嘧啶或者2,3,5,6-四氢-1h-咪唑并[1,2-a]咪唑和1-甲基-2,3,5,6-四氢咪唑并[1,2-a]咪唑。特别优选的混合物是1,5,7-三氮杂双环[4.4.0]癸-5-烯和7-甲基-1,5,7-三氮杂双环[4.4.0]癸-5-烯的混合物。
实施例:
[0063]
对于其他实施例,下列缩写具有下列含义:
[0064]
dpta:二亚丙基三胺(3,3'-二氨基二丙基胺)
[0065]
dptu:1-(3-氨基丙基)四氢嘧啶-2(1h)-酮
[0066]
tbd:1,5,7-三氮杂双环[4.4.0]癸-5-烯
[0067]
me-tbd:7-甲基-1,5,7-三氮杂双环[4.4.0]癸-5-烯
[0068]
dpta与co2的反应:
[0069]
实施例1:
[0070]
将40ml dpta的50重量%甲醇溶液转移至装备有顶置式搅拌器和气体入口的0.3l实验室高压釜中。在室温下将该高压釜用二氧化碳(5巴)吹扫3次。然后将压力再次增至5巴并开启搅拌器。在20分钟内分批注入二氧化碳以产生8-10巴的压力。然后将高压釜加热至180℃。在达到180℃时,通过进一步注入二氧化碳而将压力增至61巴。在达到61巴时,将反
应混合物搅拌16小时。然后将反应混合物冷却至室温并将该高压釜减压至环境压力。通过气相色谱法分析反应混合物。
[0071]
反应混合物(无溶剂)的组成以面积百分数给出并且为如下:
[0072]
11%dptu
[0073]
6%tbd
[0074]
20%1-甲基四氢嘧啶-2(1h)-酮
[0075]
27%1-乙基四氢嘧啶-2(1h)-酮
[0076]
实施例2:
[0077]
将40ml dpta转移至装备有顶置式搅拌器和气体入口的0.3l实验室高压釜中。在室温下将该高压釜用二氧化碳(3巴)吹扫3次。然后将压力再次增至5巴并开启搅拌器。在20分钟内分批注入二氧化碳以产生20巴的压力。然后将高压釜加热至180℃。在达到180℃时,通过进一步注入二氧化碳而将压力增至61巴。在达到62巴时,将反应混合物搅拌16小时。然后将反应混合物冷却至室温并将该高压釜减压至环境压力。通过气相色谱法分析反应混合物。
[0078]
反应混合物(无溶剂)的组成以面积百分数给出并且为如下:
[0079]
31%dptu
[0080]
6%tbd
[0081]
16%1-甲基四氢嘧啶-2(1h)-酮
[0082]
22%1-乙基四氢嘧啶-2(1h)-酮
[0083]
实施例3:
[0084]
将40ml dpta的50重量%n-甲基吡咯烷酮溶液转移至装备有顶置式搅拌器和气体入口的0.3l实验室高压釜中。在室温下将该高压釜用二氧化碳(3巴)吹扫3次。然后将压力再次增至5巴并开启搅拌器。在20分钟内分批注入二氧化碳以产生15巴的压力。然后将该高压釜加热至180℃。在达到180℃时,通过进一步注入二氧化碳而将压力增至61巴。在达到64巴时,将反应混合物搅拌16小时。然后将反应混合物冷却至室温并将该高压釜减压至环境压力。通过气相色谱法分析反应混合物。
[0085]
反应混合物(无溶剂)的组成以面积百分数给出并且为如下:
[0086]
31%dptu
[0087]
5%tbd
[0088]
13%1-甲基四氢嘧啶-2(1h)-酮
[0089]
20%1-乙基四氢嘧啶-2(1h)-酮
[0090]
dpta与碳酸二甲酯的反应
[0091]
实施例4:
[0092]
向装备有回流冷凝器的1000ml三颈烧瓶中加入100g(0.76mol)dpta和6.9g(0.05mol)tbd。通过使用冰浴将该混合物冷却至8℃。2小时后滴加75g(0.83mol,1.09当量)碳酸二甲酯。然后将反应混合物温热至20℃。在达到20℃之后用油浴将该混合物加热至90℃并在该温度下保持6小时。然后用蒸馏桥代替回流冷凝器并在大气压力和达130℃的油浴温度下蒸除产生的甲醇。在蒸馏之后将该溶液冷却至20℃并在不进一步提纯下原样用于接下来的步骤。
[0093]
示例性产物混合物组成:
[0094]
反应混合物(无溶剂)的组成以面积百分数给出并且为如下:
[0095]
78%dptu
[0096]
11%dpta
[0097]
6%tbd
[0098]
步骤ii和iii:
[0099]
实施例5:
[0100]
1-(3-氨基丙基)四氢嘧啶-2(1h)-酮(dptu)气相环化成1,5,7-三氮杂双环[4.4.0]癸-5-烯(tbd)
[0101][0102]
垂直设置具有油加热、在底部的石英玻璃料、在顶部与泵连接的液态和气态进料入口和经由转子流量计测量的气态进料(n2)的长1000mm、直径40mm的双壁玻璃反应器并将出口(底部)连接于收集烧瓶。废气连接于实验室通风橱出口。将玻璃管从上到下放入该反应器的中央并将柔性热电偶引入该管中。该反应器填充3层。首先将200ml由钢丝网构成的拉西环(直径5mm)装载于石英玻璃料上。然后引入催化剂(100ml,3mm裂片/挤出物)。该催化剂床高度为约80mm。在该催化剂床之上装载700ml拉西环,其用作液体进料和气体进料的蒸发器和加热区。
[0103]
将该管式反应器加热至反应温度(253℃)。
[0104]
dptu以来自如实施例4所公开的碳酸二甲酯与dpta在催化量的tbd下反应的粗溶液(85面积%dptu,10面积%dpta,2面积%tbd)使用。
[0105]
将如实施例4所述得到的粗dptu溶于溶剂中(10-25重量%粗dptu)并将所得溶液直接供于该反应器中的蒸发器床顶部。在1巴下从蒸发器床顶部向下通过该反应器将氮气供入该反应器中。该dptu溶液通过加热和被氮气不断夹带的合并作用而蒸发并且该蒸发器床用作该dptu/氮气料流的加热器。
[0106]
使dptu、溶剂和氮气的合并气流在该催化剂床上通过并在收集烧瓶中冷凝。有规律地从该烧瓶中取出液态样品。
[0107]
通过气相色谱法测定该液态样品的组成,给出主组分tbd和me-tbd的面积百分数。
[0108]
表1:
[0109][0110]
催化剂a是二氧化硅掺杂的氧化铝,3mm裂片
[0111]
催化剂b是无定形氧化铝,3mm挤出物
[0112]
实施例6:
[0113]
使用具有电加热的长770mm、壁厚3mm且内径12mm钢制反应器。该反应器填充3层。首先将该反应器用3-5个丝网环(3mm)和5ml玻璃球填充,然后用实施例5的催化剂a(90ml,3mm裂片)或滑石环(70ml,4
×
3.5
×
2mm)填充,然后再次用5ml玻璃球和3-5个丝网环(3mm)填充。经由泵加入包含粗dptu和该溶剂的原料溶液并使用油热蒸发器在255℃下蒸发。在该蒸发器之前将氮气引入该装置中。将该反应器的出口管加热至70-100℃并连接于收集容器。可以在该反应器的出口之后加入另一进料(通常是溶剂)以稀释该产物进料。
[0114]
将该反应器加热至反应温度(至多350℃)。
[0115]
将如实施例4所述得到的粗dptu溶于溶剂中(20-25重量%)并直接供入该装置的蒸发器中。使dptu、溶剂和氮气的合并气流在该催化剂床上通过,在该反应器之后加入额外溶剂并在收集烧瓶中冷凝产物混合物。有规律地从该烧瓶中取出液态样品。
[0116]
通过气相色谱法测定该液态样品的组成,给出主组分的面积百分数。
[0117]
表2:使用催化剂a的结果
[0118][0119][0120]
与表1相比,表2的结果表明可以通过反应压力控制式i化合物的形成。更高的压力导致形成烷基化胍类。
[0121]
表3:使用滑石环(4
×
3.5
×
2mm)的结果
[0122]
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。