一种用于2,4-d检测的人工抗体的制备方法
技术领域
1.本发明涉及材料科学领域,特别是涉及一种用于2,4-d检测的fe3o4纳米粒子人工抗体的制备方法。
背景技术:
2.2,4-d即2,4-二氯苯氧乙酸,是一种低毒性有机溶剂,分子式为c8h6cl2o3,是一种生长素类似物,用作植物生长调节剂,可广泛应用于除草剂;在普通的植物生长素定量法中显示有高的活性,但在采用植物生长素标准定量法的燕麦伸长试验中,其效颇低,影响植物代谢,因而被较多地应用于提高水果的品质和延长保鲜时间等方面。
3.2,4-d,由于其挥发性低,水溶性也较低,在自然界很难直接降解,易在水体、土壤及果蔬中残留,进而转移至生物体内;2,4-d是一种内分泌干扰物,可以通过皮肤或呼吸系统进入体内,在生物体内积累,积累到一定量时具有致畸性,同时,它还有潜在的致癌,致突变性,影响免疫及生殖系统的部分功能,若使用不当,2,4-d易引起人畜不同程度的毒副危害。因此,建立对2,4-d的残留量的精准检测具有重要意义。
4.现有的传统检测2,4-d的方法有elisa,气相色谱法、液相色谱法、气相质谱联用法等。2013年戴晓虎等人公开发明专利(cn 103149302 a)“一种土壤中2,4-d二氯苯氧乙酸的检测方法”。该方法检测步骤如下:(a)将土壤样本封干后提取土壤内容,以丙酮、水作为提取剂提取半小时提取液;(b)重复上述提取步骤得到二次提取液,离心并合并两次提取液,旋转蒸发将丙酮蒸至近干;(c)将30ml二氯甲烷加入上述步骤得到的溶液中进行萃取得到的萃取液,再将30ml二氯甲烷加入上述步骤萃取后剩余的水溶液进行二次萃取后得到的二次萃取液,将两次萃取液合并、蒸干定容;(d)采用液相色谱法测定上步蒸干定容后的溶液进行检测溶液中2,4-d浓度。2015年邹慧玲等人公开发明专利(cn 104880518 b)“一种检测叶面肥料中2,4-二氯苯氧乙酸的方法”。该发明将2,4-d标准液配成一系列已知浓度的2,4-d溶液,将该2,4-d溶液进行高效液相色谱分析,得到系列浓度2,4-d的色谱峰面积,根据峰面积得其标准曲线及线性回归方程;将待测样品处理后进行高效液相色谱分析,得到样品中2,4-d的峰面积,将峰面积代入上述线性回归方程后得到相应的2,4-d浓度,从而实现对2,4-d的检测。
5.上述elisa、液相色谱法、气相色谱法、气相质谱联用法等方法,虽然灵敏度较高,但是采用大型仪器,成本较高,选择性较差,因此,有必要寻求一种便捷和高选择性的识别目标分析物的方法,分子印迹聚合物独特的构异性,因此对目标分子具有高选择性。当前,有些团队对其开展了广泛的研究,yoko nomura等人(analytical letters,2019, 31(6):973-980)提出了通过分子印迹制备了一种对2,4-d有选择性的合成聚合物,并检查了2,4-d印迹聚合物的结合特性。使用甲基丙烯酸作为功能单体实现了对2,4-d的选择性。le sheng等人(talanta,2017, 174, 725-732)提出了通过表面引发的可逆加成断裂链转移(si-raft)聚合反应制备超顺磁性核-壳分子印迹聚合物纳米颗粒(mip),用于选择性识别实际样品中的2,4-d。成功完成了具有50nm mip层的均匀核-壳结构的构建,这有利于质量转移
并导致快速的识别动力学。静态平衡实验表明,fe3o4@mip具有令人满意的吸附容量和压印效率。此外,与结构类似物相比,fe3o4@mip对2,4-d表现出高选择性和亲和性。制备的fe3o4@mip纳米颗粒用于自来水和大白菜样品中2,4-d的选择性富集。结合rp-hplc,大白菜样品中2,4-d的回收率为93.1%至103.3%,相对标准偏差为1.7-5.4%(n = 3)。这项工作为制造结构良好的核-壳mip纳米颗粒提供了一种通用方法,可用于快速富集和高选择性分离真实样品中的目标分子。
6.综上所述,分子印迹技术可以实现高效选择性识别,其原理是当模板分子与聚合物单体接触时会形成多重作用点,通过聚合过程这种作用就会被记忆下来,当模板分子去除后,聚合物就形成了与模板分子空间构型相匹配的具有多重作用点的空穴,空穴对模板分子及类似物具有选择性识别特性,所制备的洗脱目标分子后的聚合物可称之为人工抗体。
7.为了提高对目标分析物高灵敏度的识别和专一性的检测,通常生物发光使用抗体、酶等作为分子识别材料,然而,抗体、酶等生物分子易受环境影响,价格昂贵且使用条件十分苛刻,因此,寻求一种新的目标分析物信号输出方法是目前亟待解决的问题。化学发光免疫分析法是将具有高灵敏度的化学发光测定技术与高特异性的免疫反应相结合,用于各种抗原、抗体、激素、酶等的检测分析技术。marina m. vdovenko等人(food chemistry,2013, 141(2): 865-868.)建立了2,4-d直接竞争酶联免疫吸附实验。通过改变酶联2,4-d和过氧化物酶与2,4-d结合物的浓度,优化了酶联免疫吸附性能,采用过氧化物酶催化氧化鲁米诺的化学发光法测定了结合物的酶活性。采用3-(10吩噻嗪基)丙烷-1-磺酸钠和4-吗啡啉吡啶的混合物作为化学发光信号增强剂。 检测下限为1.5 ng/ml, ic
50
为64.0 ng/ml,工作范围为6.5 ~ 545 ng/ml。 cl elisa从10的恢复值飙升的样本桔子(n = 5)和柑橘(n = 5)种植在温室使用2,4-d和包含不同的2,4
ꢀ‑ꢀ
d浓度(10
ꢀ‑ꢀ
300 ng / ml)范围从104%到92,这表明在缺乏水果提取感兴趣的基体效应。 对在越南购买的5个橘子和5个柑橘的果皮进行2,4-d含量测定,结果表明,柑橘果实中2,4-d含量(79-104
µ
g/kg)显著高于橘子(1.66
‑ꢀ
2.82
µ
g/kg)。化学发光灵敏度高,线性范围宽,但是选择性差,免疫分析是直接将化学发光底物或酶类物质标记在抗原或抗体上,通过抗原与抗体特异性结合形成的复合物而建立的一种分析方法。将化学发光与免疫分析相结合起来,可称为化学发光免疫分析,该方法具有高灵敏度,低成本,速度快等优点。
8.本发明主要制备对2,4-d具有特异性识别能力的分子印迹聚合物微球,通过化学发光免疫分析对2,4-d进行痕量检测,这就克服了化学发光的选择性差的问题,也为分子印迹识别2,4-d提供了识别信号,同时未见报道通过以2,4-d为目标分子,对其印迹制备微球,通过化学发光免疫分析法检测2,4-d。本发明以fe3o4纳米粒子为芯,聚合物为印记壳层,洗脱了位于壳层的2,4-d印迹分子,壳层的内部形成具有与印迹分子结构、大小和功能基互补的空穴结构,洗脱印迹分子的印记壳层具有对2,4-d分子的特异性识别位点,人工抗体的壳层识别位点处的氮原子提供孤对电子对与进入识别位点的目标分析物2,4-d分子中羧基中氢原子二者通过非共价键形式相互作用,实现对目标分析物分子选择性识别和检测。
技术实现要素:
9.发明目的:本发明为了弥补现有技术的不足,发明了一种用于2,4-d检测的fe3o4纳
米粒子人工抗体的制备方法,本方法具有制备步骤简单,灵敏性强,选择性高,特异性强,结合位点多,可重复使用,成本低廉的优点。
10.本发明是通过如下技术方案实现的:一种用于2,4-d检测的fe3o4纳米粒子人工抗体的制备方法,其特征在于:所述的人工抗体是以fe3o4纳米粒子为芯,聚合物为印记壳层,洗脱了位于壳层的2,4-d印迹分子,壳层的内部形成具有与印迹分子结构、大小和功能基互补的空穴结构,洗脱印迹分子的印记壳层具有对2,4-d分子的特异性识别位点,人工抗体的壳层识别位点处的氮原子提供孤对电子对与进入识别位点的目标分析物2,4-d分子中羧基中氢原子二者通过非共价键形式相互作用,实现对目标分析物分子选择性识别和检测,所述的芯-壳型fe3o4纳米粒子人工抗体的制备过程包括如下四个步骤:第一步是fe3o4纳米粒子的制备:首先,称取0.8 ~ 0.9g fecl3·
6h2o及0.2 ~ 0.4g fecl2·
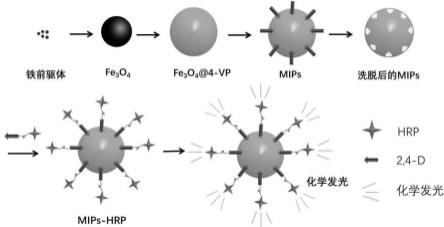
4h2o,置于含有20 ~ 30ml聚乙二醇的100ml单颈烧瓶中,以400 ~ 500rpm的转速搅拌,得到聚乙二醇混合溶液;再称取0.2 ~ 0.4g 二水柠檬酸三钠及1.4 ~ 1.5g无水乙酸钠,分别将其放入10 ~ 20 ml乙二醇溶液的50ml的单颈烧瓶中,以400 ~ 500rpm的转速搅拌,混合均匀后,将所得乙二醇混合溶液快速倒入聚乙二醇混合溶液中,搅拌1 ~ 2h后,将所得的液体倒进100ml反应釜中,在200 ~ 210℃干燥箱中恒温反应9 ~ 10h,反应结束后,将反应釜冷却至25℃,取出反应釜内物料,清洗,最后将所得fe3o4纳米粒子重新分散在去离子水中,通氮气密封保存备用;第二步是芯-壳型fe3o4纳米粒子人工抗体的制备:量取80 ~ 90ml甲醇及20 ~ 30ml的fe3o4水溶液放入250ml锥形瓶中,超声分散后,随后加入50 ~ 70mg 的4-乙烯基吡啶以及20 ~ 30mg的2,4-d,将锥形瓶中的溶液在摇床上培育18 ~ 22min,添加交联剂三羟甲基丙烷三甲基丙烯酸酯和引发剂偶氮二异丁腈后,超声4 ~ 6min,通氮气10 ~ 15min,密封,置于摇床上在 60 ~ 65℃、250 ~ 300rpm的摇床中反应16 ~ 18h,进行磁性分离后,所得芯-壳型fe3o4纳米粒子用乙醇和水反复提纯,再用体积比为 7:3 的甲醇和乙酸混合溶液洗涤三次,以去除模板分子,直到经磁性分离的上清液中284nm处无紫外吸收,得到芯-壳型fe3o4纳米粒子人工抗体,再将其重新分散在乙醇中,密封保存;第三步是酶标2,4-d的制备:称取2 ~ 4mg辣根过氧化物酶溶解于1 ~ 3ml新鲜配制的乙酸盐缓冲液中,加入2 ~ 4mg高碘酸钠,室温下以300rpm的转速反应15 ~ 20min,对乙酸盐缓冲液透析2 ~ 4h,调节ph为4.5。称取2 ~ 6mg己二胺加入到1ml乙酸盐缓冲液中,室温下以300rpm的转速反应3 ~ 4h,再用naoh将辣根过氧化物酶溶液的ph调节至9.2。反应后加入0.2ml浓度为4mg
·
l-1
的硼氢化钠水溶液,搅拌20min,加入2 ~ 4mgn-羟基琥珀酰亚胺、10mg二环己基碳二亚胺,再搅拌1h在4℃下用3500d透析袋透析,得到酶标2,4-d备用;第四步是酶联免疫化学发光实验:取第二步制备的8 ~ 10ml芯-壳型fe3o4纳米粒子人工抗体、2 ~ 5mg的2,4-d及第三步制备的1 ~ 3ml酶标2,4-d于黑色96孔板中,室温下共培育2h后,在外加磁场的存在下倒掉上清液,并用tris-hcl缓冲液洗涤三次,去除多余反应物,最后加入5.2μl的30%过氧化氢与鲁米诺混合液,通过发光值的改变,实现对2,4-d的检测。
11.相对现有技术的有益效果:2019年高大明等人公开发明专利(cn 109828108 a)“一种用于咖啡因检测的人工抗体的制备方法”,公开了一种用于咖啡因检测的人工抗体的制备方法,利用化学免疫发光法,实现对酶标抗原分子痕量检测,检测限为5.2
×
10-11
mol
·
l
ꢀ‑1,应用在免疫分析技术上,具有高灵敏度、高特异性、简单快捷、易于操作和无需任何仪器设备等优点。2013年李建平等人公开发明专利(cn 103163124 a)“用分子印迹电化学发光传感器检测微量赤霉素a3的方法”,公开了一种用分子印迹电化学发光传感器检测微量赤霉素a3的方法。当待测分子赤霉素a3与电极表面的分子印迹膜上罗丹明 b 标记的赤霉素a3进行竞争取代时,罗丹明b在一定的电压下被氧化,形成氧化态的中间产物,该中间产物可以极大地诱导放大鲁米诺微弱的电化学发光信号,导致鲁米诺底液的电化学发光降低。罗丹明b与底液中的鲁米诺在金电极上的电化学发光强度变化与赤霉素a3的浓度在1.0
×
10-11
~3.0
×
10-9
mol/l 浓度范围内呈良好的线性关系,方法检出限为3.45
×
10-12
mol/l,该方法提高了灵敏度和选择性。
12.本发明首先是fe3o4纳米粒子的制备:称取0.8 ~ 0.9g fecl3·
6h2o及0.2 ~ 0.4g fecl2·
4h2o,置于含有20 ~ 30ml聚乙二醇的100ml单颈烧瓶中,以400 ~ 500rpm的转速搅拌,得到聚乙二醇混合溶液;再称取0.2 ~ 0.4g 二水柠檬酸三钠及1.4 ~ 1.5g无水乙酸钠,分别将其放入10 ~ 20 ml乙二醇溶液的50ml的单颈烧瓶中,以400 ~ 500rpm的转速搅拌,混合均匀后,将所得乙二醇混合溶液快速倒入聚乙二醇混合溶液中,搅拌1 ~ 2h后,将所得的液体倒进100ml反应釜中,在200 ~ 210℃干燥箱中恒温反应9 ~ 10h,反应结束后,将反应釜冷却至25℃,取出反应釜内物料,清洗,最后将所得fe3o4纳米粒子重新分散在去离子水中,通氮气密封保存备用;然后是芯-壳型fe3o4纳米粒子人工抗体的制备:量取80 ~ 90ml甲醇及20 ~ 30ml的fe3o4水溶液放入250ml锥形瓶中,超声分散后,随后加入50 ~ 70mg 的4-乙烯基吡啶以及20 ~ 30mg的2,4-d,将锥形瓶中的溶液在摇床上培育18 ~ 22min,添加交联剂三羟甲基丙烷三甲基丙烯酸酯和引发剂偶氮二异丁腈后,超声4 ~ 6min,通氮气10 ~ 15min,密封,置于摇床上在 60 ~ 65℃、250 ~ 300rpm的摇床中反应16 ~ 18h,进行磁性分离后,所得芯-壳型fe3o4纳米粒子用乙醇和水反复提纯,再用体积比为 7:3 的甲醇和乙酸混合溶液洗涤三次,以去除模板分子,直到经磁性分离的上清液中284nm处无紫外吸收,得到芯-壳型fe3o4纳米粒子人工抗体,再将其重新分散在乙醇中,密封保存;其次是酶标2,4-d的制备:称取2 ~ 4mg辣根过氧化物酶溶解于1 ~ 3ml新鲜配制的乙酸盐缓冲液中,加入2 ~ 4mg高碘酸钠,室温下以300rpm的转速反应15 ~ 20min,对乙酸盐缓冲液透析2 ~ 4h,调节ph为4.5。称取2 ~ 6mg己二胺加入到1ml乙酸盐缓冲液中,室温下以300rpm的转速反应3 ~ 4h,再用naoh将辣根过氧化物酶溶液的ph调节至9.2。反应后加入0.2ml浓度为4mg
·
l-1
的硼氢化钠水溶液,搅拌20min,加入2 ~ 4mgn-羟基琥珀酰亚胺、10mg二环己基碳二亚胺,再搅拌1h在4℃下用3500d透析袋透析,得到酶标2,4-d备用;最后是酶联免疫化学发光实验:取第二步制备的8 ~ 10ml芯-壳型fe3o4纳米粒子人工抗体、2 ~ 5mg的2,4-d及第三步制备的1 ~ 3ml酶标2,4-d于黑色96孔板中,室温下共培育2h后,在外加磁场的存在下倒掉上清液,并用tris-hcl缓冲液洗涤三次,去除多余反应物,最后加入5.2μl的30%过氧化氢与鲁米诺混合液,通过发光值的改变,实现对2,4-d的检测。
13.与现有技术相比,本发明的优点在于:在本发明中,我们基于分子印迹技术制备的磁性分子印迹仿生抗体,实现了对2,4-d的检测。磁性分子印迹仿生抗体尤其适合作为2,4-d的检测工具,首先,在磁性分子印迹仿生抗体中洗脱了位于壳层的2,4-d印迹分子,壳层的
内部形成具有与印迹分子结构、大小和功能基互补的空穴结构,洗脱印迹分子的微球具有对目标分析物分子的特异性识别位点,其次,将分子印迹仿生抗体与化学发光酶联免疫分析相结合,将分子印迹的特异性与化学发光的灵敏性相结合,不仅避免了从动物体内获得抗体的不人道做法,而且提高了检测过程中试剂受酸碱度及环境温度的影响,引入磁性的分子印迹仿生抗体,使分离界限更加清晰,从而达到对目标分析物检测的目的。这种对2,4-d分子具有专识性作用的磁性分子印迹仿生抗体,以纳米技术和分子印迹技术为基础、自身富含吸电子羧基基团的特性,显现出对2,4-d目标分子的高选择性、高灵敏性检测。因此,本发明所制备的一种用于2,4-d检测的磁性分子印迹仿生抗体,其具有制备步骤简单,选择性高,灵敏性强,特异性强,结合位点多,可重复使用,成本低廉等优点。
14.综上所述,本发明所得的化学发光免疫分析可用来检测2,4-d其一:上述制备的以2,4-d为印迹分子的人工抗体经洗脱后得到具有与酶标抗原相应的空穴结构,可以高选择性地识别2,4-d。
15.其二:所制备的磁性纳米材料人工抗体在外加磁场的作用下具有较强的磁性,便于分离和提纯,减少了在分离纯化过程中的损耗。
16.其三:与传统的分子印迹技术相比,本发明结合了化学发光免疫分析法,通过加入化学发光试剂、辣根过氧化酶、清洗液等,进而大大的提高反应活性,加快了抗体与抗原的结合速度,提高检测效率。
17.其四:酶标抗原进入到所制备的人工抗体的识别位点,用化学发光仪检测酶标抗原与抗体结合前后发光强度变化,实现对2,4-d的检测,该方法灵敏度高,选择性好。
附图说明
18.图1是本发明所制备的合成磁性纳米粒子仿生抗体对2,4-d检测的技术路线图。
19.图2是本发明所制备的磁性fe3o4纳米颗粒的sem图。
20.图3是本发明所制备的磁性fe3o4纳米颗粒的磁滞回线图。
21.图4是本发明所制备的磁性fe3o4纳米颗粒的xrd图。
22.图5是本发明所制备的磁性fe3o4纳米颗粒的红外光谱图。
23.图6是本发明所制备的芯-壳型表面分子印迹聚合物的sem图。
24.图7是本发明所制备的材料的红外光谱图。
25.图8是本发明在同一时间段内化学发光强度随2,4-d浓度变化图。
26.图9是本发明中2,4-d对化学发光比率的四参数方程。
27.图10是本发明hrp活化的红外光谱图(a)和改性后的hrp的红外光谱图(b)。
28.图11是本发明所制备的2,4-d聚合物的红外光谱图(a)和酶联2,4-d的红外光谱图。
29.根据附图进一步解释本发明的技术方案:图1是本发明所制备的合成磁性纳米粒子仿生抗体对2,4-d检测的技术路线图。首先,将铁的前驱体通过水热法合成四氧化三铁纳米颗粒,然后在其表面包覆4-vp壳层,并添加2,4-d模板分子,得到分子印迹的心-壳型结构,然后用相应的洗脱液对印迹分子进行洗脱,使得壳层中留下与模板分子骨架相同并具有相应的识别模板分子能力的点位,再将模板分子及酶标模板分子加入已经制备好的分子印迹聚合物中,让其产生竞争性反应,通过
一系列的纯化洗涤后,加入化学发光基地液,最后用化学发光检测仪器进行发光测量。
30.图2是本发明所制备的磁性fe3o4纳米颗粒的sem图。由图可知磁性fe3o4纳米颗粒的粒径在约为230nm,粒径分布均匀,呈圆球状且高度分散。
31.图3是本发明所制备的磁性fe3o4纳米颗粒的磁滞回线图。原本悬浮在溶剂中的磁性fe3o4能够快速被分离,且溶液变澄清,几乎无残留,说明本实施例所制备的fe3o4纳米粒子具有快速的外加磁场响应能力,且无其它非磁性杂志的出现,在外加磁场消失后,缓慢摇动,磁性fe3o4能重新悬浮在溶剂中且无团聚或不分散现象,表明所制备的磁性fe3o4在撤去外部磁场以后,磁性消失,本实施例所制备的fe3o4能够在水溶液中放置数月仍不被氧化。
32.图4是本发明所制备的磁性fe3o4纳米颗粒的xrd图。由图可知30.2
°
处产生的吸收峰位为fe3o4(220)晶面的衍射峰;35.5
°
处出现的吸收峰则为fe3o4(311)晶面的衍射峰;43.2
°
处出现的对应的吸收峰可能为fe3o4(400)晶面的衍射峰;53.5
°
处出现的对应的吸收峰峰可能为fe3o4(422)晶面的衍射峰;57.2
°
对应的峰位可能为fe3o4(511)晶面的衍射峰;62.4
°
应的峰位可能为fe3o4(440)晶面的衍射峰,强而尖锐的反射峰位表明所制备的产物结晶良好并且在xrd图谱中没有观察到杂质相,通过scherrer公式,由最强峰(311)计算得晶粒尺寸为9.01nm。
33.图5是本发明所制备的磁性fe3o4纳米颗粒的红外光谱图。由图可知590cm-1
左右出现的吸收峰为fe-o键金属四面体的伸缩振动特征吸收峰,1080cm-1
左右的吸收峰为fe-o键的倍频峰,而3430cm-1
左右的吸收峰为羟基的特征峰,2922cm-1
左右为-ch
2-的非对称伸缩震动,1455cm-1
及2970cm-1
左右分别为甲基弯曲震动及伸缩振动吸收峰,1390cm-1
左右为-ch3的振动引起的吸收峰,1623cm-1
左右的吸收峰为c-h的弯曲振动,1050cm-1
左右为伯醇的弯曲振动特征吸收。由上述分析可以看出,该磁性材料的成功合成.图6是本实施例所制备的芯-壳型表面分子印迹聚合物的sem图。由图可知2,4-d印迹的fe3o4的粒径在约为1.00um,并清晰可见心-壳结构。
34.图7是本发明所制备的材料的红外光谱图。曲线a为fe3o4的红外光谱图,曲线b为fe3o4@4-vp的红外光谱图,曲线c为2,4-d印迹fe3o4@4-vp的红外光谱图,曲线d为印迹洗脱后的fe3o4@4-vp的红外光谱图。b中1095cm-1
为碳氢的面内弯曲震动吸收峰,1415cm-1
~1458cm-1
处的吸收峰为吡啶基团中c=c双键的伸缩振动吸收峰,889cm-1
处的吸收峰为吡啶基团中c-h的面外弯曲震动吸收峰,807cm-1
左右的吸收峰归因于-c(ch3)3中c骨架的震动峰,通过对图谱中b的分析可以看出fe3o4表面壳层由4-vp及交联剂trim组成,表明4-乙烯基吡啶壳层的成功制备。曲线c中1620 cm-1
处吸收峰的增强是由于功能单体4-乙烯基吡啶中吡啶集团的缺电子n原子与模板分子中的氢原子以氢键的方式结合的缘故,可以表明2,4-d分子的成功印迹,与此同时曲线d中2922cm-1
吸收峰的消失及1095cm-1
处峰的极度衰减则说明了模板分子的成功洗脱。
35.图8是本发明在同一时间段内化学发光强度随2,4-d浓度变化图。该图为在一定浓度的luminol、h2o2、ph下,通过间接竞争法在0~14min期间每分钟检测一次化学发光强度所得到的2,4-d浓度在0.1~1000
×
10-11
下随时间变化的化学发光图谱,结果显示,提高反应时间,化学发光值不断下降直至最低,这是由于反应物料的不断消耗所致,我们在时间为0~14min内并没有得到一个凸峰,这可能是由于该反应过于快速,而该图中的0时间应为绝对时间加上仪器的操作及联动时间。
36.图9是本发明中2,4-d对化学发光比率的四参数方程。首先,取图8(a)中最大发光点,以b/b0为纵坐标、2,4-d浓度的对数值为横坐标进行四参数标准方程拟合,该方程为y=b (a-b)/[1 (x/c)d],其中a和b分别为当logx趋近于0和 ∞时b/b0渐近线的值,c为logx对应的发光强度衰减到一半时的浓度值,d和斜率相关的常数,得出其标准曲线方程为y=0.00841 096734/[1 (logx/0.52087)
1.01582
],检测下限可达到1.33
×
10-8
mg/l(在此将检测线定义为80%比率下的物质浓度)。
[0037]
图10是本发明hrp活化的红外光谱图(a)和改性后的hrp的红外光谱图(b)。由图可知,1635 cm-1
及3300 cm-1
附近的吸收主要为水的羟基作用,1415 cm-1
处为烷烃类ch的面内弯曲震动吸收峰,2950 cm-1
处的尖峰为长链烷烃中ch的伸缩振动峰,这可能是己二胺中碳链的引入所致。曲线b中1550 cm-1
处的吸收峰为酰胺中nh的面内弯曲震动吸收峰,960 cm-1
附近为醛基的ch面外弯曲震动吸收峰,与曲线a相比,此处的吸收强度减弱,及1550 cm-1
处酰胺峰的生成说明醛基化的hrp与乙二胺发生反应生成酰胺键,因此,可以说明hrp的成功改性。
[0038]
图11是本发明所制备的2,4-d聚合物的红外光谱图(a)和酶联2,4-d的红外光谱图。由图可知,660cm-1
、860cm-1
及1060cm-1
处的吸收峰分别为取代苯类的ch面外弯曲震动、1,2,4三取代的ch面外弯曲振动吸收峰及ch面内弯曲震动吸收峰,1435cm-1
及1500cm-1
处较弱的吸收峰则为取代苯中的骨架震动所引起的,1250cm-1
处的强吸收为芳香醚中=c-o-c的反伸缩震动吸收峰,由这些峰位可以判定该物质中含有2,4-d,1095cm-1
及1650cm-1
处的吸收为酰胺类及胺类的c=o伸缩振动及c-n伸缩振动吸收峰说明2,4-d通过酰胺键被成功链接到dcc一端,1385cm-1
、2870 cm-1
及2925cm-1
处的吸收峰则分别为烷烃中ch的面内弯曲震动吸收峰、ch的对称伸缩振动吸收峰及ch的反对称伸缩振动吸收峰,这可能是由于nhs中亚甲基所带来的,1410cm-1
左右较弱的吸收峰为oh的面内弯曲震动吸收峰,3200~3700cm-1
处为oh及酰胺类的伸缩振动的叠加峰,由此可以说明2,4-d共聚物(羟基琥珀酰亚胺-2,4-d脂)的成功制备,图中曲线b为最后合成的酶联2,4-d(2,4-d-hrp)的红外谱图,与曲线a相比,3400cm-1
左右的峰位发生了明显的短波移动现象,且吸收强度增强,这可能是由于活化改性后的hrp富含氨基的原因,其中n原子为电子给体,在与hrp反应过程中,hrp末端链接的碳链上的氨基与2,4-d共聚物中的氧原子发生作用,原本的成键类型发生改变,使得n原子的能态更加稳定,因此在红外谱图下显示为波数的移动,而强度的增加则可归因于该官能团增加所致,这预示着活化改性后的hrp被成功连接到2,4-d共聚物上,形成2,4-d-hrp。
具体实施方式
[0039]
第一步是fe3o4纳米粒子的制备:首先,称取0.8 ~ 0.9g fecl3·
6h2o及0.2 ~ 0.4g fecl2·
4h2o,置于含有20 ~ 30ml聚乙二醇的100ml单颈烧瓶中,以400 ~ 500rpm的转速搅拌,得到聚乙二醇混合溶液;再称取0.2 ~ 0.4g 二水柠檬酸三钠及1.4 ~ 1.5g无水乙酸钠,分别将其放入10 ~ 20 ml乙二醇溶液的50ml的单颈烧瓶中,以400 ~ 500rpm的转速搅拌,混合均匀后,将所得乙二醇混合溶液快速倒入聚乙二醇混合溶液中,搅拌1 ~ 2h后,将所得的液体倒进100ml反应釜中,在200 ~ 210℃干燥箱中恒温反应9 ~ 10h,反应结束后,将反应釜冷却至25℃,取出反应釜内物料,清洗,最后将所得fe3o4纳米粒子重新分散在去离子水中,通氮气密封保存备用;
第二步是芯-壳型fe3o4纳米粒子人工抗体的制备:量取80 ~ 90ml甲醇及20 ~ 30ml的fe3o4水溶液放入250ml锥形瓶中,超声分散后,随后加入50 ~ 70mg 的4-乙烯基吡啶以及20 ~ 30mg的2,4-d,将锥形瓶中的溶液在摇床上培育18 ~ 22min,添加交联剂三羟甲基丙烷三甲基丙烯酸酯和引发剂偶氮二异丁腈后,超声4 ~ 6min,通氮气10 ~ 15min,密封,置于摇床上在 60 ~ 65℃、250 ~ 300rpm的摇床中反应16 ~ 18h,进行磁性分离后,所得芯-壳型fe3o4纳米粒子用乙醇和水反复提纯,再用体积比为 7:3 的甲醇和乙酸混合溶液洗涤三次,以去除模板分子,直到经磁性分离的上清液中284nm处无紫外吸收,得到芯-壳型fe3o4纳米粒子人工抗体,再将其重新分散在乙醇中,密封保存;第三步是酶标2,4-d的制备:称取2 ~ 4mg辣根过氧化物酶溶解于1 ~ 3ml新鲜配制的乙酸盐缓冲液中,加入2 ~ 4mg高碘酸钠,室温下以300rpm的转速反应15 ~ 20min,对乙酸盐缓冲液透析2 ~ 4h,调节ph为4.5。称取2 ~ 6mg己二胺加入到1ml乙酸盐缓冲液中,室温下以300rpm的转速反应3 ~ 4h,再用naoh将辣根过氧化物酶溶液的ph调节至9.2。反应后加入0.2ml浓度为4mg
·
l-1
的硼氢化钠水溶液,搅拌20min,加入2 ~ 4mgn-羟基琥珀酰亚胺、10mg二环己基碳二亚胺,再搅拌1h在4℃下用3500d透析袋透析,得到酶标2,4-d备用;第四步是酶联免疫化学发光实验:取第二步制备的8 ~ 10ml芯-壳型fe3o4纳米粒子人工抗体、2 ~ 5mg的2,4-d及第三步制备的1 ~ 3ml酶标2,4-d于黑色96孔板中,室温下共培育2h后,在外加磁场的存在下倒掉上清液,并用tris-hcl缓冲液洗涤三次,去除多余反应物,最后加入5.2μl的30%过氧化氢与鲁米诺混合液,通过发光值的改变,实现对2,4-d的检测。
[0040]
实施例:第一步是fe3o4纳米粒子的制备:首先,称取0.81g fecl3·
6h2o及0.298g fecl2·
4h2o,置于含有20 ml聚乙二醇的100ml单颈烧瓶中,以400rpm的转速搅拌,得到聚乙二醇混合溶液,再称取0.294g 二水合柠檬酸三钠及1.476g无水乙酸钠,分别将其放入 20 ml乙二醇溶液的50ml的单颈烧瓶中,以450rpm的转速搅拌,混合均匀后,将所得乙二醇混合溶液快速倒入聚乙二醇混合溶液中,搅拌1h后,将所得的液体倒进100ml反应釜中,在200℃干燥箱中恒温反应10h,反应结束后,将反应釜冷却至25℃,取出反应釜内物料,清洗,将得到的fe3o4纳米粒子重新分散在去离子水中,通氮气密封保存备用;第二步是芯-壳型fe3o4纳米粒子人工抗体的制备:量取80ml甲醇及20ml的fe3o4水溶液放入250ml的锥形瓶中,超声分散后,随后加入50mg 的4-乙烯基吡啶以及20mg的2,4-d,将锥形瓶中的溶液在摇床上培育20min,添加交联剂三羟甲基丙烷三甲基丙烯酸酯和引发剂偶氮二异丁腈后,超声5min,通氮气10min,密封,置于60℃、300rpm的摇床中反应16h,进行磁性分离后,所得芯-壳型fe3o4纳米粒子用乙醇和水反复提纯,再用体积比为 7:3 的甲醇和乙酸混合溶液洗涤三次,去除模板分子,直到经磁性分离的上清液在284nm处无紫外吸收,得到芯-壳型fe3o4纳米粒子人工抗体,再将其重新分散在乙醇中,密封保存;第三步是酶标2,4-d的制备:称取2mg辣根过氧化物酶溶解于1ml新鲜配制的乙酸盐缓冲液中,加入2mg高碘酸钠,室温下以300rpm的转速反应20min,对乙酸盐缓冲液透析2h,调节ph为4.5。称取2mg己二胺加入到1ml乙酸盐缓冲液中,室温下以300rpm的转速反应4h,再用naoh将辣根过氧化物酶溶液的ph调节至9.2。反应后加入0.2ml浓度为4mg
·
l-1
的硼氢化钠水溶液,搅拌20min,加入2mg n-羟基琥珀酰亚胺、10mg二环己基碳二亚胺,再搅拌1h
在4℃下用3500d透析袋透析,得到酶标2,4-d备用;第四步是酶联免疫化学发光实验:取第二步制备的8mg芯-壳型fe3o4纳米粒子人工抗体、2 mg的2,4-d及第三步制备的1ml酶标2,4-d于黑色96孔板中,室温下共培育2h后,在外加磁场的存在下倒掉上清液,并用tris-hcl缓冲液洗涤三次,去除多余反应物,最后加入5.2μl的30%过氧化氢与鲁米诺混合液,通过发光值的改变,实现对2,4-d的检测。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。