1.本发明涉及肿瘤细胞检测技术领域,特别涉及金掺杂共价有机框架材料及制备方法和应用、共价有机框架纳米酶生物探针及应用、试剂盒。
背景技术:
2.肿瘤转移是导致肿瘤复发和难以根治的重要原因。肿瘤的微转移(micrometastasis)是指少数具有转移潜能的肿瘤细胞脱离原发灶,转移到血液、淋巴结、骨髓或远处器官中。目前临床上的检测方法很难发现这种微转移。循环肿瘤细胞(circulating tumor cells,ctcs)是自发或因诊疗操作,由实体瘤或转移病灶释放进入外周血循环的肿瘤细胞,是恶性肿瘤患者出现术后复发或远处转移的重要标志。ctcs是公认的肿瘤转移的先导标志,其可作为肿瘤分期的重要判定标准。此外,肿瘤患者ctcs数据可作为肿瘤分期的重要预测因子和病情改善与否的标志。ctcs的早期检测有助于早期发现肿瘤的微转移、监测术后复发、评估疗效及预后或选择合适的个体化治疗。而且ctcs检测样本为血液,而血液标本易于收集且为微创性,即使是在无转移的情况下,ctcs也可作为潜在性实时标志物用于监测疾病进展和指导治疗方案的建立,预测治疗的有效性和必要性。因此,检测ctcs在医学上具有重要的意义。
3.基于磁性纳米颗粒的免疫亲和材料由于具有易操作、易修饰、对目标物分离的高特异性和高灵敏度特点,在ctcs的分离富集应用广泛。通常,生物探针一般由磁性纳米颗粒和特异性识别抗体组成,特异性识别抗体通过静电引力、范德华力、亲水相互作用力等非共价力结合在磁性纳米颗粒表面,固载的特异性识别抗体易脱落,结构稳定性差,进而影响对ctcs的检测准确性和灵敏度。
技术实现要素:
4.有鉴于此,本发明目的在于提供金掺杂共价有机框架材料及制备方法和应用、共价有机框架纳米酶生物探针及应用。本发明提供的金掺杂共价有机框架材料以及通过金硫键偶联的巯基修饰的特异性识别生物材料形成的纳米酶生物探针结构均具有优异的结构稳定性,对循环肿瘤细胞的灵敏度高。
5.为了实现上述发明目的,本发明提供以下技术方案:
6.本发明提供了一种金掺杂共价有机框架材料,包括共价有机框架材料和负载在所述共价有机框架材料上的金纳米粒子;
7.所述共价有机框架材料具有式i所示结构的重复结构单元:
[0008][0009]
所述式i中
“…”
表示连接所述重复结构单元。
[0010]
优选的,所述金纳米粒子的负载量为10~40wt%。
[0011]
本发明提供了上述技术方案所述金掺杂共价有机框架材料的制备方法,包括以下步骤:
[0012]
将三(四苯甲醛基)磷、三(4-氨基苯基)胺、乙酸和卤代芳香烃-醇溶剂混合,进行席夫碱反应,得到共价有机框架材料;
[0013]
将所述共价有机框架材料、金离子源、还原剂和醇类溶剂混合,进行还原反应,得到金掺杂共价有机框架材料。
[0014]
本发明提供了一种共价有机框架纳米酶生物探针,包括上述技术方案所述的金掺杂共价有机框架材料和与所述金掺杂共价有机框架材料通过金硫键结合的巯基修饰的特异性识别生物材料。
[0015]
优选的,所述巯基修饰的特异性识别生物材料包括巯基修饰的适配体。
[0016]
本发明提供了上述技术方案所述的共价有机框架纳米酶生物探针在制备特异性检测循环肿瘤细胞的试剂盒中的应用。
[0017]
本发明提供了一种特异性检测循环肿瘤细胞的试剂盒,包括上述技术方案所述的共价有机框架纳米酶生物探针和叶酸修饰的磁性纳米材料。
[0018]
优选的,所述叶酸修饰的磁性纳米材料包括叶酸修饰的氨基化四氧化三铁。
[0019]
优选的,所述试剂盒还包括底物,所述底物包括荧光检测底物或紫外-可见检测底物。
[0020]
优选的,所述荧光检测底物包括葡萄糖、亚铁离子和mil-53(al);
[0021]
所述紫外-可见检测底物包括对硝基苯酚和硼氢化钠。
[0022]
本发明提供了一种金掺杂共价有机框架材料,包括共价有机框架材料和负载在所
述共价有机框架材料上的金纳米粒子。本发明采用的金掺杂共价有机框架材料中,具体式i所示结构的共价有机框架材料具有有序排列的活性反应位点,较大比表面积,良好的结构稳定性,共价有机框架材料中的三苯基膦与金纳米粒子间通过共价键(配位键)结合,结构稳定性高,且同时具有良好的模拟硝酸还原酶活性和葡萄糖氧化酶活性。
[0023]
本发明提供了上述技术方案所述金掺杂共价有机框架材料的制备方法,本发明提供的制备方法操作简单,原料来源广且成本低,适宜工业化生产。
[0024]
本发明提供了一种共价有机框架纳米酶生物探针,包括上述技术方案所述的金掺杂共价有机框架材料和与所述金掺杂共价有机框架材料通过金硫键结合的巯基修饰的特异性识别生物材料。本发明利用巯基修饰的特异性识别生物材料可以特异性识别结合循环肿瘤细胞的特点,与金掺杂共价有机框架材料中的金通过au-sh共价键结合,构建了一个用于检测循环肿瘤细胞的共价有机框架纳米酶生物探针,该纳米酶生物探针具有优异的结构稳定性,具有检测方便、成本低、高灵敏性、高精确度以及高通量等优点。
[0025]
本发明提供了一种共价有机框架纳米酶生物探针,包括上述技术方案所述的金掺杂共价有机框架材料和与所述金掺杂共价有机框架材料通过金硫键结合的巯基修饰的特异性识别生物材料。本发明提供的纳米酶生物探针中,金掺杂共价有机框架材料中的金纳米粒子能够与特异性识别的巯基修饰生物材料中的巯基通过au-sh共价键结合,形成的共价有机框架纳米酶生物探针具有优异的稳定性,对循环肿瘤细胞(ctcs)的检测精确度高、灵敏度高、选择性高、检测范围宽、检测时间短、检测效率高以及检测成本低的特点。
[0026]
本发明提供了一种特异性检测循环肿瘤细胞的试剂盒,包括上述技术方案所述的共价有机框架纳米酶生物探针和叶酸修饰的磁性纳米材料。本发明提供的试剂盒检测ctcs时,ctcs膜表面过表达的叶酸受体(fr)与叶酸修饰的磁性纳米材料相互作用,使得叶酸修饰的磁性纳米材料实现对ctcs进行分离和富集。共价有机框架纳米酶生物探针具有特异性识别、捕获ctcs和信号放大的作用,而且具有高的模拟硝酸还原酶活性和葡萄糖氧化酶活性以及非生理条件下的结构稳定,能够催化底物发生还原反应或氧化反应,从而实现对ctcs的快速准确检测。而且,本发明提供的特异性检测循环肿瘤细胞的试剂盒对循环肿瘤细胞的检测精确度高、灵敏度高、检测范围宽、检测限低,成本低,具有重要的科学意义和临床应用价值。如实施例测试结果所示,本发明提供的特异性检测循环肿瘤细胞的试剂盒对循环肿瘤细胞的线性检测范围为50~20000cells/ml,检测限为17cells/ml。
[0027]
进一步的,本发明提供的所述试剂盒还包括底物,所述底物包括荧光检测底物或紫外-可见检测底物;所述荧光检测底物包括葡萄糖、亚铁离子和mil-53(al);所述紫外-可见检测底物包括对硝基苯酚和硼氢化钠。本发明提供的试剂盒能够实现紫外-可见以及荧光双信号检测,具体的,能够催化对硝基苯酚转化为对硝基苯胺,实现紫外-可见模式检测;还能够在催化葡萄糖的同时产生过氧化氢将fe
2
氧化为fe
3
,通过离子交换将mil-53(al)转化为mil-53(fe),从而猝灭mil-53(al)的荧光,实现荧光模式检测,从而实现对ctcs的快速、准确、高灵敏度检测,在肿瘤的鉴别诊断中具有潜在的应用前景。
附图说明
[0028]
图1为cof@au-apt的制备以及基于cof@au-apt检测肿瘤细胞的流程图;
[0029]
图2为实施例1制备的tpp-cof、cof@au的红外光谱图;
[0030]
图3为实施例1中fa、fe3o4、fe3o
4-nh2和fe3o
4-nh
2-fa的红外光谱图;
[0031]
图4为实施例1制备的tpp-cof、cof@au、fe3o4、mil-53(al)的tem图,其中,(a)和(b)为cofs;(c)和(d)为cofs@au;(e)为mil-53(al);(f)为fe3o4;
[0032]
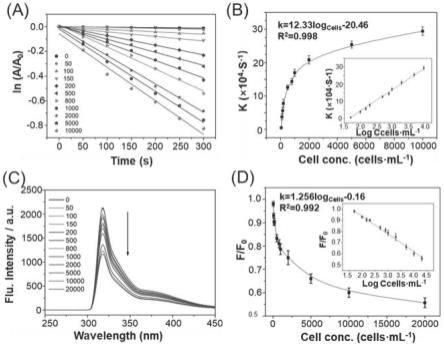
图5为cof@au-apt对肿瘤细胞选择性检测及定量检测结果图,其中,(a)和(c)为不同细胞浓度下紫外与荧光变化;(b)和(d)为紫外与荧光标准曲线。
具体实施方式
[0033]
本发明提供了一种金掺杂共价有机框架材料,包括共价有机框架材料和负载在所述共价有机框架材料上的金纳米粒子;
[0034]
所述共价有机框架材料具有式i所示结构的重复结构单元:
[0035][0036]
所述式i中
“…”
表示连接所述重复结构单元。
[0037]
在本发明中,所述金纳米粒子的负载量优选为10~40wt%,更优选为20~40wt%。在本发明中,所述金纳米粒子大部分负载在所述共价有机框架材料的表面,少量的负载在所述共价有机框架材料的孔隙结构中。
[0038]
在本发明中,所述金掺杂共价有机框架材料的制备方法,包括以下步骤:
[0039]
将三(4-甲酰苯基)胺、三(4-羟基苯基)膦、乙酸和卤代芳香烃-醇溶剂混合,进行席夫碱反应,得到共价有机框架材料;
[0040]
将所述共价有机框架材料、金离子源、还原剂和醇类溶剂混合,进行还原反应,得到金掺杂共价有机框架材料。
[0041]
在本发明中,若无特殊说明,所有的原料组分均为本领域技术人员熟知的市售商品。
[0042]
在本发明中,所述金掺杂共价有机框架材料的制备路线如图1所示。
[0043]
本发明将三(四苯甲醛基)磷、三(4-氨基苯基)胺、乙酸和卤代芳香烃-醇溶剂混合,进行席夫碱反应,得到共价有机框架材料。
[0044]
在本发明中,所述三(四苯甲醛基)磷与三(4-氨基苯基)胺的质量比优选为1:0.5~2,更优选为1:0.8~1.5,进一步优选为1:1~1.2。在本发明中,所述乙酸优选以乙酸水溶液形式使用,所述乙酸水溶液的浓度优选为1~5mol/l,优选为2~4mol/l,进一步优选为3mol/l。在本发明中,所述三(四苯甲醛基)磷质量与卤代芳香烃-醇溶剂体积的比优选为1g:50~250ml,更优选为1g:100~200ml。在本发明中,所述卤代芳香烃-醇溶剂中的卤代芳香烃优选包括邻二氯苯(o-dcb),所述醇优选包括乙醇;所述卤代芳香烃与醇的体积比优选为1:0.5~2,更优选为1:1~1.5。本发明对于所述卤代芳香烃-醇溶剂的用量没有特殊限定,能够保证席夫碱反应的顺利进行即可;在本发明的具体实施例中,所述三(四苯甲醛基)磷质量与卤代芳香烃-醇溶剂体积的比优选为1g:300ml。
[0045]
本发明对于所述混合的方式没有特殊限定,采用本领域技术人员熟知的混合方式能够将原料混合均匀即可,具体如超声混合;所述超声混合的温度优选为室温,所述超声混合的时间优选为3~10min,更优选为5~8min。
[0046]
所述混合后,本发明优选还包括将所述混合得到的悬浊液依次进行循环脱气,所述循环脱气优选包括依次进行冷冻、抽气和解冻;所述冷冻的温度优选为-10~-50℃,更优选为-50℃,所述冷冻的时间优选为6~12h,更优选为8~10h;所述解冻的温度优选为20~40℃,更优选为25~30℃;所述循环脱气的次数优选为1~5次,更优选为3~4次。
[0047]
在本发明中,所述席夫碱反应的温度优选为100~150℃,更优选为110~140℃,进一步优选为120~130℃;所述席夫碱反应的时间优选为48~96h,更优选为60~85h,进一步优选为70~80h。
[0048]
所述席夫碱反应后,本发明优选还包括将所述席夫碱反应得到的反应液进行固液分离,将得到的固体产物洗涤后干燥,得到共价有机框架材料。本发明对于所述固液分离的方式没有特殊限定,采用本领域技术人员熟知的固液分离即可,具体如过滤。在本发明中,所述洗涤优选包括依次进行邻二氯苯洗涤、丙酮洗涤和乙醚洗涤;所述邻二氯苯洗涤、丙酮洗涤和乙醚洗涤的次数独立地优选为1~5次,更优选为3~4次。在本发明中,所述干燥的温度优选为40~80℃,更优选为50~60℃,所述干燥的时间优选为6~12h,更优选为8~10h;所述干燥的方式优选为真空干燥。
[0049]
得到共价有机框架材料后,本发明将所述共价有机框架材料、金离子源、还原剂和醇类溶剂混合,进行还原反应,得到金掺杂共价有机框架材料。
[0050]
在本发明中,所述金离子源优选包括氯金酸。在本发明中,所述共价有机框架材料和金离子源中金的质量比优选为1:0.1~0.5,更优选为1:0.2~0.45,进一步优选为1:0.2~0.3。在本发明中,所述还原剂优选包括nabh4;所述还原剂优选以还原剂醇溶液形式使用,所述还原剂醇溶液中的醇优选包括甲醇;所述还原剂醇溶液的浓度优选为1~4mol/l,更优选为2~3mol/l。在本发明中,所述共价有机框架材料的质量与还原剂的物质的量的比优选为1g:0.02~0.08mol,更优选为1g:0.03~0.06mol,进一步优选为1g:0.04~0.05mol。在本发明中,所述醇类溶剂优选包括甲醇;本发明对于所述醇类溶剂的用量没有特殊限定,能够保证还原反应顺利进行即可;在本发明的具体实施例中,所述共价有机框架材料的质量与醇类溶剂的体积的比优选为1g:1l。
[0051]
在本发明的具体实施例中,所述混合优选为将共价有机框架材料和金离子源分散于醇类溶剂中搅拌,在得到的混合物中滴加还原剂醇溶液。在本发明中,所述搅拌的温度优选为室温,所述搅拌的时间优选为12~48h,更优选为20~30h。
[0052]
在本发明中,所述还原反应的温度优选为室温,所述还原反应的时间优选为1~4h,更优选为2~3h。
[0053]
所述还原反应后,本发明优选还包括将所述还原反应得到的反应液进行固液分离,将得到的固体产物洗涤后干燥,得到金掺杂共价有机框架材料。本发明对于所述固液分离的方式没有特殊限定,采用本领域技术人员熟知的固液分离即可,具体如离心分离。在本发明中,所述洗涤优选包括依次进行甲醇洗涤和水洗涤;所述甲醇洗涤和水洗涤的次数独立地优选为1~5次,更优选为3~4次。在本发明中,所述干燥的温度优选为40~80℃,更优选为50~60℃,所述干燥的时间优选为6~12h,更优选为8~10h;所述干燥的方式优选为真空干燥。
[0054]
本发明提供了一种共价有机框架纳米酶生物探针,包括上述技术方案所述的金掺杂共价有机框架材料和与所述金掺杂共价有机框架材料通过金硫键结合的巯基修饰的特异性识别生物材料。
[0055]
在本发明中,所述巯基修饰的特异性识别生物材料优选包括巯基修饰的适配体;所述巯基修饰的适配体优选包括sh-egfr,其核苷酸序列如seq id no.1所示:5'-tga atg ttg ttt ttt ctc ttt tct ata gta-3'。
[0056]
本发明提供了上述技术方案所述共价有机框架纳米酶生物探针的制备方法,优选包括以下步骤:将金掺杂共价有机框架材料、巯基修饰的适配体和磷酸盐缓冲溶液混合,进行偶联反应,得到共价有机框架纳米酶生物探针。
[0057]
在本发明中,所述共价有机框架纳米酶生物探针的制备路线如图1所示。
[0058]
在本发明中,所述金掺杂共价有机框架材料的质量与巯基修饰的适配体的物质的量的比优选为1g:1~4mmol,更优选为1g:2~3mmol。
[0059]
在本发明中,所述磷酸盐缓冲溶液(pbs)的ph值优选为7.0~7.5,更优选为7.2~7.4。在本发明中,所述金掺杂共价有机框架材料的质量与磷酸盐缓冲溶液的体积的比优选为1g:1~4l,更优选为1g:2~3l。
[0060]
在本发明中,所述巯基修饰的适配体优选以巯基修饰的适配体溶液形式使用,所述巯基修饰的适配体溶液中的溶剂优选包括pbs溶液,所述pbs溶液的ph值优选为7.0~7.5,更优选为7.2~7.4;所述巯基修饰的适配体溶液的浓度优选为50~200μmol/l,更优选为100~150μmol/l。
[0061]
在本发明的具体实施例中,所述混合优选为将金掺杂共价有机框架材料超声分散于磷酸盐缓冲溶液中,在得到的金掺杂共价有机框架材料分散液中加入巯基修饰的适配体溶液搅拌混合。在本发明中,所述超声分散的温度优选为室温,所述超声分散的时间优选为3~10min,更优选为5~8min;本发明对于所述超声分散的功率没有特殊限定,能够超声分散均匀即可。在本发明中,所述搅拌混合的温度优选为室温,本发明对于所述搅拌混合的速度和时间没有特殊限定,能够将原料混合均匀即可。
[0062]
在本发明中,所述偶联反应的温度优选为室温,所述偶联反应的时间优选为2~8h,更优选为4~6h。在本发明中,所述偶联反应过程中,巯基修饰的适配体通过巯基与金纳
米粒子之间形成配位键,使得巯基修饰的适配体偶联到金掺杂共价有机框架材料的金纳米离子表面。
[0063]
所述偶联反应后,本发明优选还包括将所述偶联反应得到的反应液进行固液分离,得到的固体产物为共价有机框架纳米酶生物探针。本发明对于所述固液分离的方式没有特殊限定,采用本领域技术人员熟知的固液分离即可,具体如离心分离。
[0064]
本发明提供了上述技术方案所述的共价有机框架纳米酶生物探针在制备特异性检测循环肿瘤细胞的试剂盒中的应用。
[0065]
本发明提供了一种特异性检测循环肿瘤细胞的试剂盒,包括上述技术方案所述的共价有机框架纳米酶生物探针和叶酸修饰的磁性纳米材料。
[0066]
在本发明中,所述共价有机框架纳米酶生物探针优选以共价有机框架纳米酶生物探针溶液形式使用,所述共价有机框架纳米酶生物探针溶液的浓度优选为0.1~0.5g/l,更优选为0.1~0.3g/l;所述共价有机框架纳米酶生物探针溶液中的溶剂优选为pbs溶液,所述pbs溶液的ph值优选为7.0~7.5,更优选为7.2~7.4。
[0067]
在本发明中,所述叶酸修饰的磁性纳米材料包括叶酸修饰的氨基化四氧化三铁(fe3o
4-nh
2-fa);所述叶酸修饰的磁性纳米材料优选以叶酸修饰的磁性纳米材料水分散液形式使用,所述叶酸修饰的磁性纳米材料水分散液的浓度优选为0.1~0.5g/l,更优选为0.1~0.3g/l。
[0068]
在本发明中,所述叶酸修饰的氨基化四氧化三铁的制备方法优选包括以下步骤:
[0069]
将氨基化四氧化三铁、叶酸、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、n-羟基琥珀酰亚胺和二甲基甲酰胺混合,进行酰胺化反应,得到叶酸修饰的氨基化四氧化三铁。
[0070]
在本发明中,所述氨基化四氧化三铁优选以氨基化四氧化三铁溶液形式使用。在本发明中,所述氨基化四氧化三铁的制备方法优选包括以下步骤:将氯化铁、乙酸铵、柠檬酸钠和乙二醇混合,进行溶剂热反应,得到四氧化三铁;将所述四氧化三铁、氨丙基三乙氧基硅、乙醇水溶液和冰醋混合,在保护气氛下进行氨基化,得到氨基化四氧化三铁溶液。
[0071]
本发明将氯化铁、乙酸铵、柠檬酸钠和乙二醇混合,进行溶剂热反应,得到四氧化三铁。
[0072]
在本发明中,所述氯化铁、乙酸铵和柠檬酸钠的摩尔比优选为1:0.5~2:2~8.5,更优选为1:1~1.5:4~6。在本发明中,所述氯化铁的物质的量与乙二醇的体积的比优选为1mol:0.5~2l,更优选为1mol:1~1.5l。在本发明中,所述混合的温度优选为30~120℃,更优选为50~100℃;所述混合的时间优选为0.5~2h,更优选为1~1.5h;所述混合的方式优选为搅拌混合。
[0073]
在本发明中,所述溶剂热反应的温度优选为120~250℃,更优选为150~200℃;所述溶剂热反应的时间优选为20~48h,更优选为24~30h;所述溶剂热反应优选在特氟隆内衬的高压反应釜中进行。
[0074]
所述溶剂热反应后,本发明优选还包括将所述溶剂热反应得到的反应液冷却至室温后固液分离,将得到的固体产物洗涤后干燥,得到四氧化三铁。本发明对于所述冷却的方式没有特殊限定,采用本领域技术人员熟知的冷却方式即可,具体如自然冷却。在本发明中,所述固液分离的方式优选为磁铁吸附分离。在本发明中,所述洗涤优选包括依次进行水洗和乙醇洗;所述水洗和乙醇洗的次数独立地优选为1~5次,更优选为3~4次。在本发明
中,所述干燥的温度优选为40~80℃,更优选为50~60℃,所述干燥的时间优选为6~12h,更优选为8~10h。
[0075]
得到四氧化三铁后,本发明将所述四氧化三铁、氨丙基三乙氧基硅、乙醇水溶液和冰醋混合,在保护气氛下进行氨基化,得到氨基化四氧化三铁溶液。
[0076]
在本发明中,所述四氧化三铁与氨丙基三乙氧基硅(aptes)的质量比优选为1:0.5~2.5,更优选为1:1~2。在本发明中,所述四氧化三铁的质量与乙醇水溶液的体积的比优选为1g:60~270ml,更优选为1g:100~200ml。在本发明中,所述乙醇水溶液中乙醇和水的体积比优选为1:0.5~2,更优选为1:1~1.5。本发明对于所述冰醋酸的用量没有特殊限定,能够将体系的ph值调节至3~5即可,所述ph值更优选为4。
[0077]
在本发明的具体实施例中,所述混合优选为将四氧化三铁加入到乙醇水溶液中,加入冰醋酸调节ph值至3~5,超声分散,然后加入氨丙基三乙氧基硅。在本发明中,所述超声分散的温度优选为室温,所述超声分散的时间优选为5~20min,更优选为10~15min;本发明对于所述超声分散的功率没有特殊限定,能够超声分散均匀即可。
[0078]
本发明对于所述保护气氛没有特殊限定,采用本领域技术人员熟知的保护气氛即可,具体如氮气或氩气。在本发明中,所述氨基化的温度优选为40~80℃,更优选为50~60℃;所述氨基化的时间优选为6~12h,更优选为8~10h。
[0079]
得到氨基化四氧化三铁溶液后,本发明将所述氨基化四氧化三铁溶液、叶酸、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐、n-羟基琥珀酰亚胺和二甲基甲酰胺混合,进行酰胺化反应,得到叶酸修饰的氨基化四氧化三铁。
[0080]
在本发明中,所述氨基化四氧化三铁溶液中氨基化四氧化三铁与叶酸的质量比优选为1:0.2~1,更优选为1:0.5~0.8。在本发明中,所述氨基化四氧化三铁与1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(edc)的质量比优选为1:0.2~0.8,更优选为1:0.4~0.6。在本发明中,所述氨基化四氧化三铁与n-羟基琥珀酰亚胺(nhs)的质量比优选为1:0.1~0.5,更优选为1:0.2~0.3。在本发明中,所述氨基化四氧化三铁的质量与二甲基甲酰胺(dmf)的体积的比优选为1g:5~25ml,更优选为1g:10~15ml。本发明对于所述混合的方式没有特殊限定,采用本领域技术人员熟知的混合方式能够将原料混合均匀即可,具体如搅拌混合。在本本发明中,所述酰胺化反应的温度优选为室温,时间优选为4~12h,更优选为5~10h;所述酰胺化反应过程中,叶酸中的羧基与氨基化四氧化三铁中的氨基形成酰胺键。
[0081]
所述酰胺化反应后,本发明优选还包括将所述酰胺化反应得到的反应液进行固液分离,将得到的固体产物洗涤后干燥,得到四氧化三铁。在本发明中,所述固液分离的方式优选为磁铁吸附分离。在本发明中,所述洗涤优选为水洗和/或乙醇洗;所述洗涤的次数优选为2~5次,更优选为3~4次。在本发明中,所述干燥的温度优选为40~80℃,更优选为50~60℃,所述干燥的时间优选为6~12h,更优选为8~10h;所述干燥的方式优选为真空干燥。
[0082]
在本发明中,所述试剂盒优选还包括底物,所述底物优选包括荧光检测底物或紫外-可见检测底物。在本发明中,所述荧光检测底物优选包括葡萄糖、亚铁离子和mil-53(al)。在本发明中,所述葡萄糖优选以葡萄糖水溶液形式使用,所述葡萄糖水溶液的浓度优选为1~5mmol/l,更优选为2~3mmol/l。在本发明中,所述亚铁离子优选来源于氯化亚铁(fecl2)和/或硫酸亚铁(feso4);所述亚铁离子优选以亚铁离子水溶液形式使用,所述亚铁
离子水溶液的浓度优选为1~5mmol/l,更优选为3~5mmol/l。在本发明中,所述紫外-可见检测底物优选包括对硝基苯酚(4-np)和硼氢化钠;所述对硝基苯酚优选以对硝基苯酚水溶液形式使用,所述对硝基苯酚水溶液的浓度优选为0.1~0.5mmol/l,更优选为0.2~0.3mmol/l;所述硼氢化钠优选以硼氢化钠水溶液形式使用,所述硼氢化钠水溶液的浓度优选为0.1~0.5mmol/l,更优选为0.1~0.3mmol/l。
[0083]
在本发明中,所述mil-53(al)的制备方法优选包括以下步骤:将水溶性铝盐、对二苯甲酸和水混合,进行水热反应后活化,得到mil-53(al)。
[0084]
在本发明中,所述水溶性铝盐优选包括硝酸铝、氯化铝和硫酸铝中的一种或几种,更优选为九水合硝酸铝。在本发明中,所述水溶性铝盐中铝与对二苯甲酸的质量比优选为0.3~2:1~6,更优选为0.5~1.5:2~5。在本发明中,所述对二苯甲酸与水的质量比优选为1g:5~35ml,更优选为1g:10~20ml。本发明对于所述混合的方式没有特殊限定,采用本领域技术人员熟知的混合方式能够将原料混合均匀即可,具体如搅拌混合。
[0085]
在本发明中,所述水热反应的温度优选为200~250℃,更优选为220~230℃;所述水热反应的时间优选为48~96h,更优选为60~80h。
[0086]
所述水热反应后,本发明优选还包括将所述水热反应得到的反应液冷却至室温后固液分离,将得到的固体产物水洗后干燥,得到mil-53(al)前驱体。本发明对于所述冷却的方式没有特殊限定,采用本领域技术人员熟知的冷却方式即可,具体如自然冷却。本发明对于所述固液分离的方式没有特殊限定,采用本领域技术人员熟知的固液分离方式即可,具体如过滤或离心分离。在本发明中,所述水洗的次数优选为3~5次,更优选为3~4次。在本发明中,所述干燥的方式优选为冷冻干燥,所述干燥的温度优选为-10~-50℃,更优选为-50℃,所述干燥的时间优选为6~12h,更优选为8~10h。
[0087]
在本发明中,所述活化的温度优选为300~400℃,更优选为330~350℃;所述活化的时间优选为48~96h,更优选为60~80h;所述活化优选在马弗炉中进行。
[0088]
所述活化后,本发明优选还包括将所述活化得到的产物置于有机溶剂中回流处理后干燥,得到mil-53(al)。在本发明中,所述回流处理优选包括依次进行第一回流处理和第二回流处理。在本发明中,所述第一回流处理用有机溶剂优选包括二甲基甲酰胺,所述第一回流处理的时间优选为6~12h,更优选为8~10h;所述第一回流处理的目的是除去未反应的对二苯甲酸。在本发明中,所述第二回流用有机溶剂优选包括乙醇;所述第二回流的时间优选为6~12h,更优选为8~10h;所述第一回流处理-第二回流处理的重复次数优选为2~3次,以彻底清除未反应的金属离子和mil-53(al)框架内的配体。在本发明中,所述干燥的温度优选为100~200℃,更优选为120~150℃;所述干燥的时间优选为6~24h,更优选为10~15h。
[0089]
在本发明中,所述试剂盒优选还包括洗涤液,所述洗涤液优选包括pbs溶液,所述pbs溶液的ph值优选为7.0~7.5,更优选为7.1~7.4。
[0090]
下面将结合本发明中的实施例,对本发明中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0091]
以下实施例中所使用的化学试剂和溶剂均为分析纯级。mcf-7来源于中国科学院
昆明动物研究所细胞库。
[0092]
实施例1
[0093]
(1)制备金掺杂共价有机框架材料(cofs@au)
[0094]
将10mg三(四苯甲醛基)磷、10mg三(4-氨基苯基)胺、2.4ml邻二氯苯(o-dcb)、0.6ml乙醇和0.12ml浓度为3mol/l的乙酸加入至耐压反应管中,在室温条件下超声分散5min,将得到悬浊液冷冻-抽气-解冻循环脱气3次,然后在120℃条件下席夫碱反应72h,反应完毕后过滤分离,将得到的固体产物依次采用o-dcb、丙酮和乙醚分别洗涤3次,在60℃条件下真空干燥至恒重,得到共价有机框架材料(tpp-cof)。
[0095]
将10mg tpp-cof和4mg haucl4·
4h2o分散在10ml甲醇中,在室温条件下搅拌24h,得到混合物;将0.2ml浓度为2mol/l的nabh4甲醇溶液在搅拌条件下滴加至上述混合物中,滴加完毕后继续搅拌反应2h,反应完毕后离心分离,将得到的固体产物用甲醇和去离子水分别洗涤3次,在60℃条件下真空干燥至恒重,得到cofs@au。
[0096]
(2)制备共价有机框架纳米酶生物探针(cofs@au-apt)
[0097]
将1mg cofs@au加入至2mlpbs溶液中(ph 7.4)中,在室温条件下超声分散5min,加入20μl浓度为100μmol/l的sh-egfr(其核苷酸序列如seq id no.1所示:5'-tga atg ttg ttt ttt ctc ttt tct ata gta-3')的pbs溶液至分散液中,在室温条件下搅拌4h,离心分离,得到的固体产物为cofs@au-apt。将cofs@au-apt分散于5mlpbs溶液中,待用。
[0098]
(3)制备fe3o
4-nh
2-fa
[0099]
将2.70g六水合氯化铁、7.70g乙酸铵和0.8g柠檬酸钠加入到140ml乙二醇中,在100℃条件下搅拌1h,转移至200ml特氟隆内衬的高压反应釜中,在200℃条件下溶剂热反应24h,反应完毕后将高压反应釜自然冷却至室温,利用磁铁将产物分离,用去离子水洗涤3次,乙醇洗涤3次,在在60℃条件下冷冻干燥10h,得到fe3o4。
[0100]
将750mg fe3o4加入到100ml乙醇水溶液(乙醇/水体积比=1:1)中,用冰醋酸调节ph值至4.0,在室温条件下超声分散10min,加入1ml aptes混合均匀,在60℃、n2保护条件下氨基化10h,得到fe3o
4-nh2。
[0101]
在50ml dmf中加入40mg fe3o
4-nh2、20mg fa、16mg edc和9mg nhs,在室温条件下搅拌8h,用磁铁收集固体产物,用乙醇洗涤3次,在50℃条件下真空干燥10h,得到fe3o
4-nh
2-fa。
[0102]
(4)制备mil-53(al)
[0103]
将1300mg al(no3)3·
9h2o和288mg对二苯甲酸超声分散于5ml的去离子水中,置于30ml特氟隆内衬的高压反应釜中,在220℃条件下水热反应72h;反应结束后冷却至室温,过滤,将得到的固体产物超纯水洗涤3次后在-50℃条件下冷冻干燥10h,得到mil-53(al)前驱体。将mil-53(al)前驱体置于马弗炉中在330℃条件下活化72h,在dmf中回流8h,然后在乙醇中回流8h,重复上dmf回流-乙醇回流2次,在120℃条件下干燥12h,得到mil-53(al)。
[0104]
图1为cof@au-apt的制备以及基于cof@au-apt检测肿瘤细胞的流程图。由图1所可知,通过肿瘤细胞膜表面过表达的叶酸受体(fr)与cof@au-apt相互作用,利用fe3o
4-nh
2-fa分离ctcs,cofs@au作为捕获和信号放大的探针,能够构建紫外-可见及荧光双模式ctcs特异性富集分离的细胞传感平台,在肿瘤的鉴别诊断中具有潜在的应用前景。
[0105]
图2为tpp-cof、cof@au的红外光谱图。由图2可知,峰值出现在1705cm-1
、3460cm-1
和3337cm-1
分别归属于三(四苯甲醛基)磷(tapp)和三(4-氨基苯基)胺(tfpa)的c=o和n-h伸缩振动特征峰;在1617cm-1
处出现了一个明显的c=n伸缩振动峰,表明,cofs是由氨基和醛基之间通过席夫碱反应连接而形成的。
[0106]
图3为fa、fe3o4、fe3o
4-nh2和fe3o
4-nh
2-fa的红外光谱图。由图3可知,fe3o4的特征吸收峰分别出现在586cm-1
和3420cm-1
处,分别与fe-o键和-oh的伸缩振动对应。fe3o
4-nh2出现在2973cm-1
、2885cm-1
和1050cm-1
的吸收峰是由官能团化后aptes中的c-h和si-o-si键的伸缩振动和不对称弯曲引起的。此外fe3o
4-nh
2-fa中1706cm-1
处的吸收峰为fa中的c=o键的伸缩振动引起的,表明,fa在fe3o4表面成功修饰。
[0107]
图4为tpp-cof、cof@au、fe3o4、mil-53(al)的透射电镜(tem)图,其中,(a)和(b)为cofs;(c)和(d)为cofs@au;(e)为mil-53(al);(f)为fe3o4。由图4可知,mil-53(al)的棒状结构和直径为300nm的fe3o4的球形结构清晰可见。
[0108]
实施例2
[0109]
基于cof@au-apt的肿瘤细胞双信号检测方法
[0110]
(1)紫外-可见模式检测
[0111]
将0.5ml浓度为0.1mg/ml的fe3o
4-nh
2-fa水分散液和0.5ml浓度为0.1mg/ml的cofs@au-apt水分散液分别加入至浓度分别为0、50、100、150、200、500、800、1000、2000、5000和10000cells/ml的mcf-7细胞悬液中,混合均匀后37℃条件下孵育60min。孵育结束后采用磁铁分离除去上清液并用pbs清洗三次以出去未结合的游离细胞。在收集的混合液中加入2ml浓度为0.2mmol/l的4-np水溶液和浓度为0.1mmol/l的nabh4水溶液搅拌混合均匀,离心,将所得上清液进行紫外-可见光谱分析。其中,紫外光谱分析采用时间分辨的紫外-可见光谱测定406nm处4-np的吸光度。
[0112]
(2)荧光模式检测
[0113]
将0.5ml浓度为0.1mg/ml的fe3o
4-nh
2-fa水分散液和0.5ml浓度为0.1mg/ml的cofs@au-apt水分散溶分别加入至浓度分别为0、50、100、150、200、500、800、1000、2000、5000、10000和20000cells/ml不同浓度的mcf-7细胞悬液中,混合均匀后37℃条件下孵育60min。孵育结束后采用磁铁分离除去上清液并用pbs清洗三次以出去未结合的游离细胞。在收集的混合液中加入2ml浓度为2mmol/l的葡萄糖水溶液和浓度为5mmol/l的fecl2水溶液搅拌15min后离心分离,将上清液加入到0.2ml浓度为0.1mg/ml的mil-53(al)水分散液中搅拌均匀,测试在305nm激发荧光光谱。
[0114]
图5为cof@au-apt对肿瘤细胞选择性检测及定量检测结果图,其中,(a)和(c)为不同细胞浓度下紫外与荧光变化;(b)和(d)为紫外与荧光标准曲线在不同细胞浓度(50~10000cells/ml)下采用时间依赖紫外-可见光谱测定在406nm处4-np的吸光度。ln(a/a0)之间存在线性相关关系,其中a0为4-np在406nm处的原始吸光强度,a为cof@au-apt在不同细胞浓度和不同催化还原时间下还原4-np在406nm处的最终吸光强度。催化反应速率常数k由线性斜率ln(a/a0)随时间的变化得到。反应速率常数k值随mcf-7细胞浓度的增加而增加。从50~10000cells/ml细胞浓度下,反应速率常数k值随不同浓度细胞的变化而变化。k与mcf-7细胞对数浓度的校正曲线回归方程为k=12.33log
cells-20.46,r2=0.998,检出限(lod)值为17cells/ml(s/n=3)。
[0115]
cof@au-apt在催化葡萄糖的同时产生过氧化氢将fe
2
氧化为fe
3
,通过离子交换
将mil-53(al)转化为mil-53(fe),从而猝灭mil-53(al)的荧光。mil-53(al)的荧光强度随着细胞浓度的增加而降低。f/f0与mcf-7细胞对数浓度的校正曲线的回归方程为f/f0=1.256log
cells-0.16,r2=0.992,lod值为17cells/ml(s/n=3)。
[0116]
(3)回收率
[0117]
标准添加方法被用来量化各种mcf-7细胞浓度,以评估细胞传感器的应用。将mcf-7细胞分散在人血清中,人血清中mcf-7细胞的浓度分别为200cells/ml、500cells/ml、1000cells/ml、5000cells/ml和10000cells/ml,然后用细胞传感器(cof@au-apt)检测人血清中肿瘤细胞的浓度。
[0118]
表1人血清中肿瘤细胞加标回收结果
[0119][0120]
由表1可知,本发明提供的cof@au-apt的检测回收率为98.1~104.4%,相对标准偏差为3.1~9.1%,表明,本发明提供的cof@au-apt在临床检测和诊断中具有潜在的应用价值。
[0121]
以上所述仅是本发明的优选实施方式,应当指出,对于本技术领域的普通技术人员来说,在不脱离本发明原理的前提下,还可以做出若干改进和润饰,这些改进和润饰也应视为本发明的保护范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。