1.本发明涉及一种溴虫腈的制备方法。
背景技术:
2.溴虫腈(chlorfenapyr),商品名称:除尽,化学名称:4-溴-2-(4-氯苯基)-1-乙氧基甲基-5-(三氟甲基)吡咯-3-腈(m.p.100℃~101℃),为苯基吡咯类新型杀虫杀螨剂,其作用点为昆虫细胞内线粒体,不同于目前常用的杀虫剂。该产品具有杀虫谱广、活性高、持效期长、对有益生物安全和对环境友好等特点,具有触杀、胃毒作用。
3.溴虫腈的合成方法较多,其中2-对氯苯基-5-三氟甲基-3-氰基吡咯是溴虫腈合成的关键中间体,该中间体经溴代反应、再与氯甲基乙基醚发生取代反应,即可制得溴虫腈。然而由于2-对氯苯基-5-三氟甲基-3-氰基吡咯的吡咯环上含有氯苯基、三氟甲基和氰基,上述基团均为吸电子基团,进而导致该中间体在溴代反应时的活性较低,进而导致制得产物和纯度不高。
4.巴斯夫公司(cn 110382462 a)发现在进行溴代反应时,以dipea代替三乙胺作为缚酸剂,可提高溴代反应的收率。同时后处理更为简便,且容易回收碱dipea。所述专利实施例中a3溴代步骤的收率为88.2%。对比例b3以三乙胺作为碱时,溴代步骤的收率仅为75.3%。
5.付庆等(除尽的合成研究与工艺改进,浙江工业大学硕士学位论文)在合成4-溴-2-(4-氯苯基)-5-三氟甲基吡咯-3-腈时,以2-(4-氯苯基)-5-三氟甲基吡咯-3-腈作为原料,用以氯仿和dmf的混合溶剂,液溴作为溴化试剂,在催化剂m存在下,进行溴代反应,产物的纯度最高达93.6%,收率最高可达99%。产生含hbr 15%以上的废水,可通氯气氧化以回收溴素。虽然该反应的收率较高,但是并未公开所述m的具体组成,并且产物纯度也不高。
6.白若飞等(cn 102746208 a)将2-(4-氯苯基)-5-三氟甲基吡咯-3-腈和氧化剂加入到极性溶剂中,混合均匀后,加入溴素,反应完成后,经后处理得到4-溴-2-(4-氯苯基)-5-三氟甲基吡咯-3-腈,收率92%-96%。
7.刘源等(cn 109369498 a)公开了将2-对氯苯基-5-三氟甲基吡咯-3-腈溶液与溴素溶液同时连续通入到微反应器中进行反应,所得混合溶液在微反应器中流经两个以上反应模块后与通入到微反应器中的双氧水溶液进行反应,蒸馏,得到4-溴-2-对氯苯基-5-三氟甲基吡咯-3-腈。
8.徐尚成等(南京农业大学学报,2004,27(2):105~108)公开了在碳酸氢钠作为缚酸剂,使2-对氯苯基-5-三氟甲基吡咯-3-腈溶液与溴素发生溴代反应,收率为91.6%。
9.陆阳等(现代农药,2007,6(2):22-25和28)公开了2-对氯苯基-5-(三氟甲基)吡咯-3-腈在氯苯中与溴发生取代反应,得到2-对氯苯基-4-溴-5-(三氟甲基)吡咯-3-腈,反应过程中无需加入缚酸剂,收率为90.7%。
10.现有技术中制备溴虫腈的过程中,为了提高溴代反应的收率和纯度,开展了大量的研究,然而得到产物收率和纯度仍然有待提高,并且需要降低生产成本。因此,开发新的
制备溴虫腈的方法仍然具有巨大的市场前景和社会效益。
技术实现要素:
11.本发明所要解决的技术问题是:提供了一种溴虫腈的制备方法,其产物纯度高,收率高,反应条件温和,且后处理简单。
12.为解决该技术问题,本发明提供了一种制备溴虫腈的方法,其包含如下步骤:
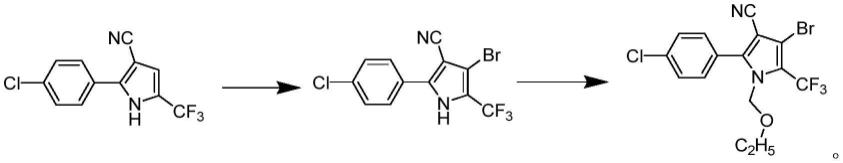
13.1)在混合溶剂中,在催化剂和缚酸剂存在下,将2-对氯苯基-5-三氟甲基吡咯-3-腈与溴素发生溴代反应,制得中间体4-溴-2-对氯苯基-5-三氟甲基吡咯-3-腈;
14.2)将4-溴-2-对氯苯基-5-三氟甲基吡咯-3-腈在碱的作用下分别与氯溴甲烷、乙醇钠反应,经后处理,得到目标产物溴虫腈;
15.具体的反应式如下:
[0016][0017]
优选地,步骤1)所述混合溶剂为四氢呋喃与另一种惰性溶剂的混合溶剂。
[0018]
合适的惰性溶剂是脂族烃类,优选脂族c
5-c
16
烃,更优选c
5-c
16
链烷烃,或者c
5-c
16
环烷烃,如戊烷、己烷、环己烷或石油醚;卤代烃类,优选卤代脂族c
1-c6链烷烃,或者卤代芳族c
6-c
10
烃类,如ch2cl2、chcl3、ccl4、ch2clch2cl、ccl3ch3、chcl2ch2cl、ccl2ccl2或氯苯;醚类,优选c
1-c6环烷基醚、c
1-c6烷基-c
1-c6烷基醚、c
1-c6烷基-c
3-c6环烷基醚、c
1-c6多元醇-c
1-c6烷基醚和c
1-c6烷基-c
6-c
10
芳基醚,如ch3ch2och2ch3、(ch3)2choch(ch3)2、ch3oc(ch3)3(mtbe)、ch3och3(dme)、ch3och2ch2och3、ch3oc(ch3)2ch2ch3和二甘醇;酯类,优选脂族c
1-c6醇类与脂族c
1-c6羧酸的酯、芳族c
6-c
10
醇类与芳族c
6-c
10
羧酸的酯、ω-羟基-c
1-c6羧酸的环状酯,如ch3c(o)och2ch3、ch3c(o)och3、ch3c(o)och2ch2ch2ch3、ch3c(o)och(ch3)ch2ch3、ch3c(o)oc(ch3)、ch3ch2ch2c(o)och2ch3、ch3ch(oh)c(o)och2ch3、ch3ch(oh)c(o)och3、ch3c(o)och2ch(ch3)2、ch3c(o)och(ch3)2、ch3ch2c(o)och3、苯甲酸苄基酯和γ-丁内酯;碳酸酯类,如碳酸亚乙酯、碳酸亚丙酯、ch3ch2oc(o)och2ch3和ch3oc(o)och3;腈类,优选c
1-c6腈类,如acn和ch3ch2cn;醇类,优选c
1-c4醇类和c
2-c4链烷二醇,如ch3oh、ch3ch2oh、ch3ch2ch2oh、ch3ch(oh)ch3、ch3(ch2)3oh和c(ch3)3oh、ch2(oh)ch2(oh)、ch3ch(oh)ch2oh;酰胺类和脲衍生物,优选dmf、nmp、dma、dmi、dmpu、hmpa;此外还有dmso和环丁砜。
[0019]
更优选地,步骤1)所述混合物溶剂为四氢呋喃和四氯化碳的混合溶剂;最优选地,所述混合溶剂中,四氢呋喃和四氯化碳的体积比为5:1-2。
[0020]
优选地,步骤1)所述的催化剂为铁粉、二氧化锰或三溴化铁;更优选为铁粉。
[0021]
优选地,步骤1)所述的缚酸剂选自4-二甲氨基吡啶、dipea、三乙胺、碳酸氢钠、碳酸氢钾、氢氧化钠、氢氧化钾、碳酸钠、碳酸钾;最优选地,所述缚酸剂选自4-二甲氨基吡啶。
[0022]
优选地,步骤1)所述的2-对氯苯基-5-三氟甲基吡咯-3-腈、溴素、催化剂和缚酸剂的摩尔比为1:1.0-2.0:0.1-0.2:1.0-3.0,更优选地,所述摩尔比为1:1.0-1.2:0.1-0.15:1.0-1.5。
[0023]
优选地,步骤1)的反应温度为30-60℃,更优选为40-50℃;
[0024]
优选地,步骤1)的反应时间为2-10小时,更优选为3-5小时。
[0025]
步骤1)的反应混合物可以以常规方式后处理,例如通过与水混合,分离各相并且合适的话,可采用常规方式提纯粗产物。通常在步骤1)反应完毕后,通过蒸馏从反应混合物除去溶剂。此外,也可以再反应完成后,向混合物中加入水,再蒸馏除去部分溶剂,然后除去水层并将含有式iii化合物的有机层干燥,例如通过加入吸湿性材料。步骤1)的产物的也可以通过从有机层结晶或者通过除去有机溶剂而得到。
[0026]
优选地,所述步骤1)的后处理方法为:待反应液冷却,加入水,搅拌,蒸馏除去混合溶剂,有大量淡黄色固体在水相中析出,过滤,滤饼用水洗涤三次,收集滤饼,干燥,得中间体4-溴-2-对氯苯基-5-三氟甲基吡咯-3-腈。
[0027]
步骤2)的反应过程可采用现有技术常见的方式进行。
[0028]
优选地,步骤2)的反应过程为:将4-溴-2-对氯苯基-5-三氟甲基吡咯-3-腈溶于四氢呋喃中,缓慢滴加含有氢化钠的四氢呋喃,搅拌反应,然后滴加溴氯甲烷,继续反应,再用含有乙醇钠的四氢呋喃溶液处理,再在反应一段时间,tlc检测反应进程,反应完毕后,降温、过滤,脱去溶剂,用正己烷/乙酸乙酯混合溶剂进行重结晶,得到白色固体溴虫腈。
[0029]
由于2-对氯苯基-5-三氟甲基-3-氰基吡咯的吡咯环上含有氯苯基、三氟甲基和氰基,上述基团均为吸电子基团,进而导致该中间体在溴代反应时的活性较低。本发明的方法通过在溴化步骤中引入催化剂,可有效降低溴化反应的活化能,进而促进反应进行提高反应收率。
[0030]
此外,由于溴化反应中会生成氢溴酸,现有技术中通常会引入缚酸剂,中和反应生成的酸,进而促使反应正向移动。本技术的发明人在考察不同的缚酸剂对于反应收率和纯度的影响时,意外地发现了以4-二甲氨基吡啶作为缚酸剂时,相对于常规的缚酸剂如三乙胺、dipea、碳酸氢钠等,反应收率可显著提高。
[0031]
再者,现有技术中付庆等(《农药》,2006,45(6):385-386、391)使用以氯仿作为主要溶剂的混合溶剂(该文献中溴化反应的纯度95%,收率98%,显著高于其他文献报道),本技术的发明人考察了不同溶剂对于产物收率和纯度的影响,发现在本发明的催化体系下,以四氢呋喃和四氯化碳作为混合溶剂时,收率和纯度最佳。
[0032]
本发明的另一方面,提供了一种溴虫腈中间体4-溴-2-对氯苯基-5-三氟甲基吡咯-3-腈的制备方法,其包含如下步骤:
[0033]
1)在混合溶剂中,在催化剂和缚酸剂存在下,将2-对氯苯基-5-三氟甲基吡咯-3-腈与溴素发生溴代反应,制得中间体4-溴-2-对氯苯基-5-三氟甲基吡咯-3-腈;
[0034]
具体的反应式如下:
[0035][0036]
优选地,步骤1)所述混合溶剂为四氢呋喃与另一种惰性溶剂的混合溶剂。
[0037]
合适的惰性溶剂是脂族烃类,优选脂族c
5-c
16
烃,更优选c
5-c
16
链烷烃,或者c
5-c
16
环烷烃,如戊烷、己烷、环己烷或石油醚;卤代烃类,优选卤代脂族c
1-c6链烷烃,或者卤代芳族c
6-c
10
烃类,如ch2cl2、chcl3、ccl4、ch2clch2cl、ccl3ch3、chcl2ch2cl、ccl2ccl2或氯苯;醚
类,优选c
1-c6环烷基醚、c
1-c6烷基-c
1-c6烷基醚、c
1-c6烷基-c
3-c6环烷基醚、c
1-c6多元醇-c
1-c6烷基醚和c
1-c6烷基-c
6-c
10
芳基醚,如ch3ch2och2ch3、(ch3)2choch(ch3)2、ch3oc(ch3)3(mtbe)、ch3och3(dme)、ch3och2ch2och3、ch3oc(ch3)2ch2ch3和二甘醇;酯类,优选脂族c
1-c6醇类与脂族c
1-c6羧酸的酯、芳族c
6-c
10
醇类与芳族c
6-c
10
羧酸的酯、ω-羟基-c
1-c6羧酸的环状酯,如ch3c(o)och2ch3、ch3c(o)och3、ch3c(o)och2ch2ch2ch3、ch3c(o)och(ch3)ch2ch3、ch3c(o)oc(ch3)、ch3ch2ch2c(o)och2ch3、ch3ch(oh)c(o)och2ch3、ch3ch(oh)c(o)och3、ch3c(o)och2ch(ch3)2、ch3c(o)och(ch3)2、ch3ch2c(o)och3、苯甲酸苄基酯和γ-丁内酯;碳酸酯类,如碳酸亚乙酯、碳酸亚丙酯、ch3ch2oc(o)och2ch3和ch3oc(o)och3;腈类,优选c
1-c6腈类,如acn和ch3ch2cn;醇类,优选c
1-c4醇类和c
2-c4链烷二醇,如ch3oh、ch3ch2oh、ch3ch2ch2oh、ch3ch(oh)ch3、ch3(ch2)3oh和c(ch3)3oh、ch2(oh)ch2(oh)、ch3ch(oh)ch2oh;酰胺类和脲衍生物,优选dmf、nmp、dma、dmi、dmpu、hmpa;此外还有dmso和环丁砜。
[0038]
更优选地,步骤1)所述混合物溶剂为四氢呋喃和四氯化碳的混合溶剂;最优选地,所述混合溶剂中,四氢呋喃和四氯化碳的体积比为5:1-2。
[0039]
优选地,步骤1)所述的催化剂为铁粉、二氧化锰或三溴化铁;更优选为铁粉。
[0040]
优选地,步骤1)所述的缚酸剂选自4-二甲氨基吡啶、dipea、三乙胺、碳酸氢钠、碳酸氢钾、氢氧化钠、氢氧化钾、碳酸钠、碳酸钾;最优选地,所述缚酸剂选自4-二甲氨基吡啶。
[0041]
优选地,步骤1)所述的2-对氯苯基-5-三氟甲基吡咯-3-腈、溴素、催化剂和缚酸剂的摩尔比为1:1.0-2.0:0.1-0.2:1.0-3.0,更优选地,所述摩尔比为1:1.0-1.2:0.1-0.15:1.0-1.5。
[0042]
优选地,步骤1)的反应温度为30-60℃,更优选为40-50℃;
[0043]
优选地,步骤1)的反应时间为2-10小时,更优选为3-5小时。
[0044]
步骤1)的反应混合物可以以常规方式后处理,例如通过与水混合,分离各相并且合适的话,可采用常规方式提纯粗产物。通常在步骤1)反应完毕后,通过蒸馏从反应混合物除去溶剂。此外,也可以再反应完成后,向混合物中加入水,再蒸馏除去部分溶剂,然后除去水层并将含有式iii化合物的有机层干燥,例如通过加入吸湿性材料。步骤1)的产物的也可以通过从有机层结晶或者通过除去有机溶剂而得到。
[0045]
优选地,所述步骤1)的后处理方法为:待反应液冷却,加入水,搅拌,蒸馏除去混合溶剂,有大量淡黄色固体在水相中析出,过滤,滤饼用水洗涤三次,收集滤饼,干燥,得中间体4-溴-2-对氯苯基-5-三氟甲基吡咯-3-腈。
[0046]
与现有技术相比,本发明的有益效果是:
[0047]
(1)本发明的方法与现有技术中制备溴虫腈的方法相比,中间体4-溴-2-对氯苯基-5-三氟甲基吡咯-3-腈的收率较高,降低了反应成本;
[0048]
(2)本发明的方法反应步骤简单,反应条件温和,同时反应中使用的试剂也均为廉价易得的试剂,易于实现工业化。
[0049]
(3)本发明的方法产物纯度高,仅需简单的后处理即可实现高纯度的产物,节省了后续的提纯步骤。
具体实施方式
[0050]
下面通过实施例来具体说明本发明的内容。在本发明中,以下实施例是为了更好
地阐述本发明,并不是用来限制本发明的范围。下述实施例中所用的材料、试剂等,如无特殊说明,均可从商业途径得到。
[0051]
实施例1
[0052][0053]
在500ml反应瓶中,加入27.1g(约0.1mol)2-对氯苯基-5-三氟甲基吡咯-3-腈,用200ml四氢呋喃和50ml四氯化碳溶解,然后加入铁粉0.6g,4-二甲氨基吡啶14.7g(约0.12mol),搅拌30min。随后缓慢滴入液溴19.8g(约0.11mol),控制温度为40℃~50℃,滴毕,室温反应4h。反应完毕后,待反应液冷却,加入水,搅拌,蒸馏除去混合溶剂,有大量淡黄色固体在水相中析出,过滤,滤饼用水洗涤三次,收集滤饼,干燥,得33.2g黄白色固体4-溴-2-对氯苯基-5-三氟甲基吡咯-3-腈,hplc检测产物纯度为98.5%,收率95.0%。
[0054]
实施例2-5不同溶剂对反应收率和产物纯度的影响。
[0055]
实施例2
[0056]
与实施例1的区别在于用250ml四氢呋喃代替200ml四氢呋喃和50ml四氯化碳,其他条件与实施例1相同。
[0057]
实施例3
[0058]
与实施例1的区别在于用250ml四氯化碳代替200ml四氢呋喃和50ml四氯化碳,其他条件与实施例1相同。
[0059]
实施例4
[0060]
与实施例1的区别在于用200ml四氯化碳和50ml四氢呋喃代替200ml四氢呋喃和50ml四氯化碳,其他条件与实施例1相同。
[0061]
实施例5
[0062]
与实施例1的区别在于用200ml四氢呋喃和50氯苯代替200ml四氢呋喃和50ml四氯化碳,其他条件与实施例1相同。
[0063]
实施例2-5的收率和纯度数据如下所示:
[0064]
实施例编号纯度(%)收率(%)实施例296.891.8实施例395.792.5实施例493.088.1实施例593.882.3
[0065]
从实施例1-5可以看出,在以四氢呋喃和四氯化碳作为混合溶剂时,产物的收率和纯度均有大幅度的提升。
[0066]
实施例6-9不同缚酸剂对反应收率和产物纯度的影响。
[0067]
实施例6
[0068]
与实施例1的区别在于用14.7g的dipea代替14.7g的4-二甲氨基吡啶,其他条件与实施例1相同。
[0069]
实施例7
[0070]
与实施例1的区别在于用14.7g的三乙胺代替14.7g的4-二甲氨基吡啶,其他条件
与实施例1相同。
[0071]
实施例8
[0072]
与实施例1的区别在于用14.7g的碳酸氢钠代替14.7g的4-二甲氨基吡啶,其他条件与实施例1相同。
[0073]
实施例9
[0074]
与实施例1的区别在于用14.7g的碳酸钠代替14.7g的4-二甲氨基吡啶,其他条件与实施例1相同。
[0075]
实施例6-9的收率和纯度数据如下所示:
[0076]
实施例编号纯度(%)收率(%)实施例695.386.3实施例793.682.7实施例894.276.3实施例992.679.8
[0077]
从实施例1和6-9可以看出,以4-二甲氨基吡啶作为缚酸剂时,产物的收率和纯度均有大幅度的提升。
[0078]
实施例10-12不同催化剂对反应收率和产物纯度的影响。
[0079]
实施例10
[0080]
与实施例1的区别在于用0.1mol的二氧化锰代替0.1mol的铁粉,其他条件与实施例1相同。
[0081]
实施例11
[0082]
与实施例1的区别在于用0.1mol的三溴化铁代替0.1mol的铁粉,其他条件与实施例1相同。
[0083]
实施例12
[0084]
与实施例1的区别在于不加入催化剂,其他条件与实施例1相同。
[0085]
实施例10-12的收率和纯度数据如下所示:
[0086]
实施例编号纯度(%)收率(%)实施例1094.588.7实施例1195.185.3实施例1293.274.7
[0087]
从实施例1和10-12可以看出,以铁粉作为催化剂时,产物的收率和纯度均有大幅度的提升。
[0088]
实施例13溴虫腈的合成
[0089]
在300ml反应瓶中,加入17.5g(0.05mol)4-溴-2-对氯苯基-5-三氟甲基吡咯-3-腈,然后加入70ml四氢呋喃,搅拌溶解,缓慢滴加含有3.0g(60%,0.075mol)氢化钠的70ml四氢呋喃中,搅拌反应,30℃下搅拌0.5h,然后滴加12.9g(0.10mol)溴氯甲烷,在60℃反应6h,再用含有7.5g(0.12mol)乙醇钠的四氢呋喃溶液处理,再在60℃反应7h,降温、过滤,脱去溶剂,用正己烷/乙酸乙酯混合溶剂进行重结晶,得到18.6g白色固体溴虫腈,收率91.3%。
[0090]
尽管已经示出和描述了本发明的实施例,对于本领域的普通技术人员而言,可以
理解在不脱离本发明的原理和精神的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由所附权利要求及其等同物限定。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。