1.本发明涉及药物化学领域,具体涉及一种奥特康唑的制备方法。
背景技术:
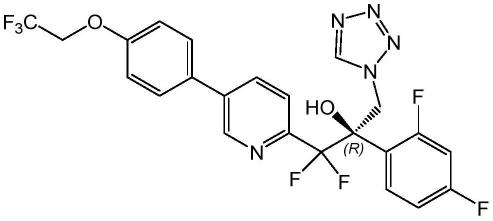
2.奥特康唑(vt-1161,英文名称:oteseconazole,cas编号:1340593-59-0),其化学名称为(r)-2-(2,4-二氟苯基)-1,1-二氟-3-(1h-四氮唑-1-基)-1-(5-(4-(2,2,2-三氟乙氧基)苯基)吡啶-2-基)丙烷-2-醇,化学结构式如式(ⅰ)所示:
[0003][0004]
奥特康唑是一款由美国mycovia公司开发的新型口服小分子选择性真菌cyp51抑制剂,且对真菌cyp51的选择性显著优于其他常用唑类抗真菌药。从目前的临床研究来看,奥特康唑也表现出了优秀的药物动力学特征、疗效以及安全性。临床上用于复发性外阴阴道念珠菌病、侵袭性真菌感染和甲真菌病等疾病的治疗。
[0005]
中国专利cn 106458900b公开了一种奥特康唑的制备方法,可由中间体1于乙酸溶剂中在原甲酸三甲酯和乙酸钠的存在下与有机叠氮试剂tmsn3(叠氮基三甲基硅烷)进行反应而制备,反应过程如工艺路线1所示。
[0006]
然而,采用tms合成vt-1161时,反应过程中容易生成甲酰胺杂质(化学结构式如式(ⅱ)所示)和乙酰胺杂质(化学结构式如式(iii)所示),而且含量较高,原料的残留也较多,因此不利于控制原料药质量,纯化过程的压力较大。此外,tms价格昂贵,商业化供应不足,且该物质闪点很低,只有75
°
f,工业化生产时极易爆炸,工艺安全风险高,不利于大规模的工业化生产。
[0007][0008]
由此可见,急需开发一种新的奥特康唑制备方法,以减少杂质的产生、提高目标产品的质量,并降低工艺的成本和安全风险,使所得的制备工艺更加适宜工业规模的生产。
技术实现要素:
[0009]
为克服现有技术中存在的上述不足,本发明的目的是提供一种奥特康唑(vt-1161)的制备方法,该制备方法可以有效减少制备过程中甲酰胺杂质和乙酰胺杂质的产生,而且工艺操作安全,易于工业应用,由此所制得的vt-1161杂质少、质量好。
[0010]
本发明提供的奥特康唑的制备方法为:以中间体1为起始原料,在无机叠氮化物和三烷基氯硅烷si(r)
3-cl的存在下进行反应,由此制得式(ⅰ)所示的vt-1161,
[0011][0012]
其中,所述无机叠氮化物为叠氮化钠或叠氮化钾,所述三烷基氯硅烷si(r)
3-cl中,r表示c1~c6的烷基。
[0013]
在以中间体1为原料制备vt-1161的过程中,本发明的发明人发现,直接使用有机叠氮试剂(例如,中国专利cn 106458900b中使用的tms)或无机叠氮化物(例如,叠氮化钠等)均无法取得理想的结果。本发明的发明人还发现,使用无机叠氮化物和三烷基氯硅烷si(r)
3-cl进行一锅法反应,反应过程中无机叠氮化物和三烷基氯硅烷原位生成叠氮基三烷基硅烷试剂,此方法则能够有效减少反应过程中甲酰胺杂质和乙酰胺杂质的产生,原料中间体1的残留也能得到明显降低,因而有利于控制目标产品的质量,提高原料的利用率。由
于副产物、残留原料等杂质的减少,后续的纯化压力也能得到大幅减轻,由此可制得高纯度的目标产品。此外,使用无机叠氮化物和三烷基氯硅烷还能够避免直接使用低闪点的叠氮基三烷基硅烷试剂,提高了制备工艺的安全性,进一步降低了生成成本,所得的制备工艺更加适用于工业化规模的生产。
[0014]
本发明提供的制备方法中,所使用的起始原料中间体1可以为市购产品,也可参考中国专利cn 106458900b进行制备。
[0015]
在根据本发明的制备方法的一些实施方式中,所述无机叠氮化物可以为叠氮化钠。
[0016]
在根据本发明的制备方法的一些实施方式中,所述三烷基氯硅烷si(r)
3-cl中的r可进一步表示c1~c4的烷基,例如,甲基、乙基等;在一些优选的实施方式中,所述三烷基氯硅烷为三甲基氯硅烷。
[0017]
在根据本发明的制备方法的一些实施方式中,所述叠氮化钠与所述中间体1的摩尔比可以为2~5:1,包括但不限于2:1、2.5:1、3:1、3.5:1、4:1、4.5:1、5:1等摩尔比值或任意的摩尔比区间;在一些优选的实施方式中,所述叠氮化钠与所述中间体1的摩尔比可以为2.5~4.5:1。
[0018]
在根据本发明的制备方法的一些实施方式中,所述叠氮化钠与所述三甲基氯硅烷的摩尔比可以为1:1~5,包括但不限于1:1、1:1.5、1:2、1:2.5、1:3、1:3.5、1:4、1:4.5、1:5等摩尔比值或任意的摩尔比区间;在一些优选的实施方式中,所述叠氮化钠与所述三甲基氯硅烷的摩尔比可以为1:1~3。
[0019]
在根据本发明的制备方法的一些实施方式中,所述制备方法的反应体系中还包括乙酸钠、原甲酸三甲酯以及包含冰乙酸的溶剂体系。
[0020]
在根据本发明的制备方法的一些实施方式中,所述溶剂体系可以选自冰乙酸、或可以选自冰乙酸与乙酸乙酯、2-丁醇中的一种或多种所组成的混合溶剂;在一些优选的实施方式中,所述溶剂体系选自冰乙酸、或选自冰乙酸与乙酸乙酯以1:0.5~2的质量比所组成的混合溶剂。
[0021]
在根据本发明的制备方法的一些实施方式中,所述溶剂体系按质量可以为所述中间体1的1.5~5倍,包括但不限于1.5、2、2.5、3、3.5、4、4.5、5等倍数值或任意的倍数区间。
[0022]
在根据本发明的制备方法的一些实施方式中,所述乙酸钠与所述中间体1的摩尔比可以为1~3:1,包括但不限于1:1、1:1.5、1:2、1:2.5、1:3等摩尔比值或任意的摩尔比区间;在一些优选的实施方式中,所述乙酸钠与所述中间体1的摩尔比可以为1~1.5:1。
[0023]
在根据本发明的制备方法的一些实施方式中,所述原甲酸三甲酯与所述中间体1的摩尔比可以为1~4:1,包括但不限于1:1、1:1.5、1:2、1:2.5、1:3、1:3.5、1:4等摩尔比值或任意的摩尔比区间;在一些优选的实施方式中,所述原甲酸三甲酯与所述中间体1的摩尔比可以为1.5~3:1。
[0024]
在根据本发明的制备方法的一些实施方式中,所述原甲酸三甲酯加入所述反应体系时的温度可以为35~65℃,加入原甲酸三甲酯时适当提高温度能够达到其反应活化能,促使反应快速发生;在一些优选的实施方式中,所述原甲酸三甲酯加入所述反应体系时的温度可以为45~55℃。
[0025]
在根据本发明的制备方法的一些实施方式中,所述反应的反应温度可以为65~85
℃;在一些优选的实施方式中,所述反应的反应温度可以为75~85℃。
[0026]
在根据本发明的制备方法的一些实施方式中,所述制备方法还可以包括以下纯化过程:所述反应结束后,使用有机溶剂萃取得到萃取液,经过碱洗、水洗后,脱干所述有机溶剂得到粗产物,所述粗产物使用异丙醇作为良溶剂、正庚烷作为不良溶剂进行重结晶。
[0027]
在一些优选的实施方式中,萃取溶剂可以使用甲基叔丁基醚、乙酸乙酯等有机溶剂。
[0028]
本发明提供的制备方法中,纯化过程中可以使用本领域常规的洗涤方法、过滤方法(例如抽滤、离心等)、干燥方法、结晶方法等。
[0029]
本发明提供的制备方法使用无机叠氮化物/三烷基氯硅烷代替叠氮基三烷基硅烷试剂一锅法合成vt-1161,能够有效控制反应过程中甲酰胺杂质和乙酰胺杂质的生成,因而有利于控制目标产品的质量,提高原料的利用率,并减轻后续纯化过程的压力。此外,本发明提供的制备方法操作简单,提高了工艺安全性,降低了生成成本。因此,本发明提供的制备方法非常适宜于大规模的工业化生产,具有重要的经济和社会价值。
具体实施方式
[0030]
以下结合具体实施例对本发明的技术方案做进一步详细说明。
[0031]
本发明的实施例和对比例中所使用的中间体1参考中国专利cn 106458900b自制,其他试剂如无特别说明,均为市售产品。
[0032]
本发明的实施例和对比例中所使用的有关物质检测方法为高效液相色谱法,其他操作方法如无特别说明,均为本领域的常规方法。
[0033]
高效液相色谱法所使用的设备和检测条件如下:
[0034]
色谱仪:agilent 1260。
[0035]
溶液配制:
[0036]
流动相a:0.05%三氟乙酸的水溶液。
[0037]
流动相b:0.05%三氟乙酸的乙腈溶液。
[0038]
稀释液:乙腈/水(1:1,v/v)。
[0039]
检测条件:
[0040][0041]
如无特别说明,本发明中所使用的百分数均为质量百分数。
[0042]
实施例1
[0043]
向反应容器中加入5g(0.06mol,1.06eq)无水乙酸钠和70ml冰醋酸,再加入35g(0.056mol)中间体1,加入10g(0.154mol,2.74eq)叠氮化钠和16g(0.147mol,2.62eq)三甲基氯硅烷,升温至45℃,加入16g(0.22mol,3.93eq)原甲酸三甲酯。
[0044]
然后升温至75-85℃,保温5小时,反应结束,取样检测反应液(检测结果如表1所示)。向反应液中加入100g水和50g甲基叔丁基醚,分层,有机层中加入100g 15%的氢氧化钠溶液,洗涤分层。然后用100g水洗涤,分层,有机层减压脱干,所得粗产物中加入70g异丙醇升温至溶解,滴加280g正庚烷,降温至20℃结晶。过滤、干燥得24.8g vt-1161,纯度:99.98%(hplc),收率83.9%,甲酰胺杂质和乙酰胺杂质均未检出。
[0045]
实施例2
[0046]
向反应容器中加入7g(0.08mol,1.43eq)无水乙酸钠和50ml冰醋酸、50ml乙酸乙酯组成的混合溶剂,再加入35g(0.056mol)中间体1,加入12g(0.18mol,3.21eq)叠氮化钠和27g(0.25mol,4.46eq)三甲基氯硅烷,升温至55℃,加入7g(0.07mol,1.25eq)原甲酸三甲酯的7ml乙酸乙酯溶液。
[0047]
然后升温至65-75℃,保温24小时,反应结束,取样检测反应液(检测结果如表1所示)。向反应液中加入100g水和200g乙酸乙酯,分层,有机层中加入100g 20%的氢氧化钾溶液,洗涤分层。然后用100g水洗涤,分层,有机层减压脱干,所得粗产物中加入100g异丙醇升温至溶解,滴加350g正庚烷,降温至0℃结晶。过滤、干燥得25.2g vt-1161,纯度:99.99%(hplc),收率85.1%,甲酰胺杂质和乙酰胺杂质均未检出。
[0048]
对比例
[0049]
参考中国专利cn 106458900b的第0487段制备得到vt-1161合成反应液,取样检测,检测结果如表1所示。
[0050]
表1反应液检测结果
[0051] 反应液纯度甲酰胺杂质乙酰胺杂质中间体1实施例197.33%0.10%0.22%0.06%实施例297.48%0.27%0.07%0.14%对比例94.61%2.01%0.35%0.73%
[0052]
通过表1结果可知,本发明提供的制备方法能够有效降低反应过程中甲酰胺杂质和乙酰胺杂质的生成,原料中间体1的残留也明显降低。
[0053]
除非特别限定,本发明所用术语均为本领域技术人员通常理解的含义。
[0054]
本发明所描述的实施方式仅出于示例性目的,并非用以限制本发明的保护范围,本领域技术人员可在本发明的范围内作出各种其他替换、改变和改进,因而,本发明不限于上述实施方式,而仅由权利要求限定。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。