1.本技术涉及分子动力学模拟技术,更具体地说,涉及一种基于材料结构模拟的分子动力学势函数的评估方法和系统。
背景技术:
2.分子动力学(molecular dynamics,md)作为材料学重要的模拟方法之一,对于模拟材料在原子尺度上的现象有不可替代的作用。对于辐照损伤、缺陷演化、相变、断裂等现象的模拟,往往需要对几十万甚至上亿个原子进行超过一百万步的模拟。在如此小的尺度下,这些现象有大量模拟细节都难以在实验中直接验证,而这些细节恰恰是揭示各种材料现象的机制、加深材料学的理论认识的重要信息。因此,如何确保这些模拟细节的准确性就变得至关重要。
3.分子动力学模拟依赖于势函数来确定模拟时原子间的相互作用,然而势函数的性能一般只由其作者在发表文献时提及,而且往往也只提及有利的方面而淡化性能较差的方面。这使得科研工作者对他们使用的某个势函数缺乏一个全面的了解和认识,对模拟结果带来了一定的不确定性。为了对势函数进行有效评估,需要使用该势函数来模拟相关结构的一些基本物理性质,然后把这些性质与实验值进行比对。这些待评估的物理性质应包括但不限于结构的内聚能、弹性系数、点缺陷形成能、表面形成能、熔点、热膨胀系数、界面能等等。当今科研界已发表的晶体势函数种类繁多,包含了单质、化合物、金属合金等物质类别,其晶体结构也主要包含了立方、四方、正交、六角等多种晶系,每一类物质和晶系都有不同数量的物理量需要进行测试,而每一种物理量所需要的结构和模拟参数都有所差异,评估模拟的工作量、复杂度和繁琐度都比较大,因此业界需要一种通用的、全面的、自动化算法流程来处理势函数的评估工作。
技术实现要素:
4.本技术要解决的技术问题在于,针对现有技术的上述缺陷,提供一种通用的、可自动化的、能够基于任意材料结构对任意势函数进行评估的基于材料结构模拟的分子动力学势函数的评估方法。
5.本技术为解决其技术问题所采用的技术方案是:提出一种基于材料结构模拟的分子动力学势函数的评估方法,包括如下步骤:
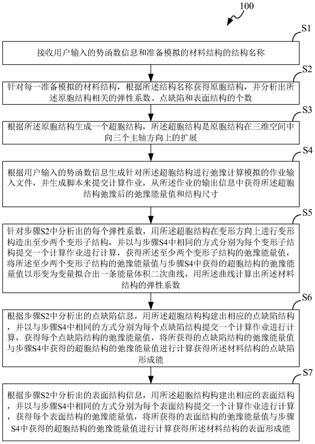
6.s1、接收用户输入的势函数信息和准备模拟的材料结构的结构名称;
7.s2、针对每一准备模拟的材料结构,根据所述结构名称获得原胞结构,并分析出所述原胞结构相关的弹性系数、点缺陷和表面结构的个数;
8.s3、根据所述原胞结构生成一个超胞结构,所述超胞结构是原胞结构在三维空间中向三个主轴方向上的扩展;
9.s4、根据用户输入的势函数信息生成针对所述超胞结构进行弛豫计算模拟的作业输入文件,并生成脚本来提交计算作业,从所述作业的输出信息中获得所述超胞结构弛豫
后的弛豫能量值和结构尺寸;
10.s5、针对步骤s2中分析出的每个弹性系数,用所述超胞结构在变形方向上进行变形构造出至少两个变形子结构,并以与步骤s4中相同的方式分别为每个变形子结构提交一个计算作业进行计算,获得所述至少两个变形子结构的弛豫能量值,将所述至少两个变形子结构的弛豫能量值与步骤s4中获得的超胞结构的弛豫能量值以形变为变量拟合出一条能量-体积二次曲线,用所述曲线根据如下公式计算出所述材料结构的弹性系数:
[0011][0012]
其中,e为弛豫能量值,v为体积,v0为步骤s4中获得的超胞结构弛豫后的弛豫结构的体积;
[0013]
s6、根据步骤s2中分析出的点缺陷信息,用所述超胞结构构建出相应的点缺陷结构,并以与步骤s4中相同的方式分别为每个点缺陷结构提交一个计算作业进行计算,获得每个点缺陷结构的弛豫能量值,将所获得的点缺陷结构的弛豫能量值与步骤s4中获得的超胞结构的弛豫能量值根据如下公式进行计算获得所述材料结构的点缺陷形成能:
[0014]edef
=en±
m-(ne0±
mμi)
[0015]
其中,e
def
为点缺陷形成能,en±m为包括m个缺陷原子的结构能量,e0为步骤s4中获得的超胞结构的每原子平均弛豫能量,n为该超胞结构中原子的数量,m为缺陷本身的原子数量,μi为第i类元素的完美结构中的原子化学势,空位缺陷计算用减号,间隙缺陷计算用加号;
[0016]
s7、根据步骤s2中分析出的表面结构信息,用所述超胞结构构建出相应的表面结构,并以与步骤s4中相同的方式分别为每个表面结构提交一个计算作业进行计算,获得每个表面结构的弛豫能量值,将所获得的表面结构的弛豫能量值与步骤s4中获得的超胞结构的弛豫能量值根据如下公式进行计算获得所述材料结构的表面形成能:
[0017][0018]
其中,e
(ijk)
是(ijk)面的表面形成能,是包含了n个原子表面结构的能量,e0为步骤s4中获得的超胞结构的每原子平均弛豫能量,a
(ijk)
是(ijk)表面的表面积。
[0019]
根据本技术所述的基于材料结构模拟的分子动力学势函数的评估方法的一个实施例中,所述步骤s1中接收的势函数信息包括势函数名称、参数值或参数文件。
[0020]
根据本技术所述的基于材料结构模拟的分子动力学势函数的评估方法的一个实施例中,所述步骤s2中根据所述结构名称获得原胞结构包括:根据所述结构名称从材料学通用结构数据库中获得所述材料结构的原胞结构。
[0021]
根据本技术所述的基于材料结构模拟的分子动力学势函数的评估方法的一个实施例中,所述步骤s3进一步包括:在所述原胞结构的任何一边长度小于势函数的截断距离的两倍时,将所述超胞结构的每个方向扩展到原胞结构尺寸的整数倍直至超越所述截断距离的两倍。
[0022]
根据本技术所述的基于材料结构模拟的分子动力学势函数的评估方法的一个实
施例中,所述步骤s3进一步包括:将所述超胞结构在每个方向的尺寸至少扩展达到
[0023]
根据本技术所述的基于材料结构模拟的分子动力学势函数的评估方法的一个实施例中,所述步骤s4进一步包括:作业提交至高性能计算服务器或超级计算系统,并且作业在提交后调用系统中的分子动力学程序来进行模拟工作。
[0024]
根据本技术所述的基于材料结构模拟的分子动力学势函数的评估方法的一个实施例中,所述步骤s5进一步包括:
[0025]
s51、对于每个弹性系数,用所述超胞结构在变形方向上分别作1%、0.5%、-0.5%、-0.1%的拉伸或剪切形变,构造出四个变形子结构;
[0026]
s52、以与步骤s4中相同的方式分别为四个变形子结构提交计算作业进行计算,获得四个变形子结构的弛豫能量值;
[0027]
s53、将所述四个变形子结构的弛豫能量值与步骤s4中获得的超胞结构的弛豫能量值以形变为变量拟合出一条能量-体积二次曲线,用曲线进行拟合计算出相应的弹性系数;
[0028]
s54、重复所述步骤s51至s53,直至步骤s2中分析出的所有弹性系数计算完毕。
[0029]
根据本技术所述的基于材料结构模拟的分子动力学势函数的评估方法的一个实施例中,所述步骤s2中分析出的所述原胞结构的点缺陷包括空位缺陷和间隙缺陷;所述步骤s6中用所述超胞结构构建出相应的点缺陷结构进一步包括:对于空位缺陷,为原胞结构内的每种元素的每种站位分别构建一个空位缺陷结构;对于间隙缺陷,为原胞结构内的每一处较大空间为每种元素分别构建一个间隙缺陷结构。
[0030]
根据本技术所述的基于材料结构模拟的分子动力学势函数的评估方法的一个实施例中,所述步骤s2中分析出的所述原胞结构的表面结构包括小指数界面;所述步骤s7中构建的表面结构的长度、宽度、厚度和真空层均超过所用势函数的截断距离的两倍。
[0031]
本技术还提出一种系统,包括处理器和存储器,所述存储器存储有计算机程序,所述计算机程序被处理器执行时实现如上所述的基于材料结构模拟的分子动力学势函数的评估方法。
[0032]
本技术还提出一种计算机可读存储介质,存储有计算机程序,所述计算机程序被处理器执行时实现如上所述的基于材料结构模拟的分子动力学势函数的评估方法。
[0033]
实施本技术的基于材料结构模拟的分子动力学势函数的评估方法,具有以下有益效果:(1)本技术的基于材料结构模拟的分子动力学势函数的评估方法是一种可以灵活评估分子动力学势函数的通用方法,该方法可以使用任意材料结构对任意势函数进行评估,对势函数的有效评估和筛选具有重大的促进作用;(2)该方法可以在最小的人工干预下,在超级计算或高性能计算平台上进行流水线式的高通量运行,实现对势函数的批量快速评估和筛选,大大减轻了科研工作者在进行材料模拟前的前期工作。
附图说明
[0034]
下面将结合附图及实施例对本技术作进一步说明,附图中:
[0035]
图1是根据本技术一个实施例的基于材料结构模拟的分子动力学势函数的评估方法的流程图;
[0036]
图2是根据本技术另一实施例的基于材料结构模拟的分子动力学势函数的评估方
法的详细流程图;
[0037]
图3是铀金属的α-u结构的原胞结构的示意图;
[0038]
图4是扩展后的α-u结构的超胞结构的示意图;
[0039]
图5是计算α-u的c
11
弹性系数的拟合曲线;
[0040]
图6是α-u的空位缺陷结构的示意图;
[0041]
图7a是α-u的一种间隙缺陷结构的示意图;
[0042]
图7b是α-u的另一种间隙缺陷结构的示意图;
[0043]
图8是α-u的(001)表面结构的示意图;
[0044]
图9是铀金属的γ-u结构的原胞结构的示意图;
[0045]
图10是扩展后的γ-u结构的超胞结构的示意图;
[0046]
图11是计算γ-u的c
11
弹性系数的拟合曲线;
[0047]
图12是γ-u的空位缺陷结构的示意图;
[0048]
图13是γ-u的间隙缺陷结构的示意图;
[0049]
图14a是γ-u的(100)表面结构的示意图;
[0050]
图14b是γ-u的(110)表面结构的示意图;
[0051]
图14c是γ-u的(111)表面结构的示意图;
[0052]
图15是根据本技术一个实施例的系统的结构示意图。
具体实施方式
[0053]
为了使本技术的目的、技术方案及优点更加清楚明白,以下结合附图及实施例,对本技术进行进一步详细说明。应当理解,此处所描述的具体实施例仅仅用以解释本技术,并不用于限定本技术。并且,在不冲突的情况下,本技术中的实施例及实施例中的特征可以相互组合。
[0054]
本技术提出一种基于材料结构模拟的分子动力学势函数的评估方法,能够在知道最少的势函数和结构信息之后,自动地使用该势函数模拟出指定结构的多种物理性质,从而达到全面和快速评估势函数的目的。图1示出了根据本技术一个实施例的基于材料结构模拟的分子动力学势函数的评估方法100的流程图。如图1所示,该基于材料结构模拟的分子动力学势函数的评估方法100包括如下步骤:
[0055]
步骤s1,接收用户输入的势函数信息和准备模拟的材料结构的结构名称。其中,用户输入的势函数信息至少包括势函数名称、参数值或参数文件。
[0056]
步骤s2,针对每一准备模拟的材料结构,根据材料结构的结构名称获得原胞结构,并分析出原胞结构相关的弹性系数、点缺陷和表面结构的个数。
[0057]
步骤s3,根据原胞结构生成一个超胞结构,所述超胞结构是原胞结构在三维空间中向三个主轴方向上的扩展。
[0058]
步骤s4,根据用户输入的势函数信息生成针对超胞结构进行弛豫计算模拟的作业输入文件,并生成脚本来提交计算作业,从所述作业的输出信息中获得所述超胞结构弛豫后的弛豫能量值和结构尺寸。
[0059]
步骤s5,针对步骤s2中分析出的每个弹性系数,用所述超胞结构在变形方向上进行变形构造出至少两个变形子结构,并以与步骤s4中相同的方式分别为每个变形子结构提
交一个计算作业进行计算,获得所述至少两个变形子结构的弛豫能量值,将所述至少两个变形子结构的弛豫能量值与步骤s4中获得的超胞结构的弛豫能量值以形变为变量拟合出一条能量-体积二次曲线,用所述曲线根据如下公式(1)计算出所述材料结构的弹性系数:
[0060][0061]
其中,e为弛豫能量值,v为体积,v0为步骤s4中获得的超胞结构弛豫后的弛豫结构的体积。
[0062]
步骤s6,根据步骤s2中分析出的点缺陷信息,用所述超胞结构构建出相应的点缺陷结构,并以与步骤s4中相同的方式分别为每个点缺陷结构提交一个计算作业进行计算,获得每个点缺陷结构的弛豫能量值,将所获得的点缺陷结构的弛豫能量值与步骤s4中获得的超胞结构的弛豫能量值根据如下公式(2)进行计算获得所述材料结构的点缺陷形成能:
[0063]edef
=en±
m-(ne0±
mμi)
ꢀꢀꢀ
(2)
[0064]
其中,e
def
为点缺陷形成能,en±m为包括m个缺陷原子的结构能量,e0为步骤s4中获得的超胞结构的每原子平均弛豫能量,n为该超胞结构中原子的数量,m为缺陷本身的原子数量,μi为第i类元素的完美结构中的原子化学势,空位缺陷计算用减号,间隙缺陷计算用加号。
[0065]
步骤s7,根据步骤s2中分析出的表面结构信息,用所述超胞结构构建出相应的表面结构,并以与步骤s4中相同的方式分别为每个表面结构提交一个计算作业进行计算,获得每个表面结构的弛豫能量值,将所获得的表面结构的弛豫能量值与步骤s4中获得的超胞结构的弛豫能量值根据如下公式(3)进行计算获得所述材料结构的表面形成能:
[0066][0067]
其中,e
(ijk)
是(ijk)面的表面形成能,是包含了n个原子表面结构的能量,e0为步骤s4中获得的超胞结构的每原子平均弛豫能量,a
(ijk)
是(ijk)表面的表面积。
[0068]
根据本技术上述实施例的基于材料结构模拟的分子动力学势函数的评估方法100可以使用任意结构对任意势函数进行评估,对势函数的有效评估和批量快速筛选具有重大的促进作用。
[0069]
图2进一步示出了根据本技术另一具体实施例的基于材料结构模拟的分子动力学势函数的评估方法200的流程图。参见图2所示,该基于材料结构模拟的分子动力学势函数的评估方法200开始于步骤s201。
[0070]
然后在步骤s202中,接收用户输入的势函数和结构信息。用户输入的势函数信息包括势函数名称、参数值或参数文件等,用户输入的结构信息至少包括准备模拟的所有结构的结构名称。
[0071]
然后该方法200针对所有输入的结构进行逐个模拟,在步骤s203中判断是否所有结构都进行了计算模拟,若是,则流程进入步骤s222结束,若否,则进入步骤s204。
[0072]
步骤s204中,根据用户输入的结构信息获取基态原胞结构。原胞结构的信息包括原胞边长、角度和所有原子的位置。具体实现时,可以根据用户输入的结构名称从材料学通
用结构数据库中获得所述材料结构的原胞结构。常用的结构数据库包括但不限于icsd(https://icsd.products.fiz-karlsruhe.de)、cod(http://www.crystallography.net/cod/)、amcsd(http://rruff.geo.arizona.edu/ams/amcsd.php)等。
[0073]
然后在步骤s205中,根据原胞结构分析出该结构相关的弹性系数、点缺陷和表面结构的个数。原胞结构的弹性系数的数量根据材料学通用知识由原胞的晶系结构所决定。例如对于立方结构,只计算c
11
、c
12
和c
44
;对于六角结构,在立方结构的基础上加上c
13
和c
33
;对于四方结构,在六角结构的基础上加上c
66
;对于正交结构,在四方结构的基础上加上c
22
、c
23
和c
55
。对于其余复杂晶系,根据材料学作相应的调整。对于最一般的结构,弹性系数最多包括21个(c
11
,c
12
,
……
,c
16
,c
22
,c
23
,
……
,c
66
),即i,j=1-6的所有c
ij
且去掉c
ij
=c
ji
的重复项。原胞结构的点缺陷包括空位缺陷和间隙缺陷。原胞结构的表面结构包括常用的小指数界面,miller index指数不超过3,例如(100)、(111)、(310)面。
[0074]
然后在步骤s206中,根据原胞结构生成一个超胞结构。所述超胞结构是原胞结构在三维空间中向三个主轴方向上的扩展。如果原胞结构的任何一边长度小于势函数的截断距离的两倍,则超胞结构在该边方向需要扩展至原胞结构尺寸的整数倍直至超越势函数的截断距离的两倍。优选地,超胞结构在每个方向的尺寸至少扩展达到
[0075]
然后在步骤s207中,根据用户输入的势函数信息生成一个作业输入文件对所述超胞结构进行弛豫计算模拟,即不限制结构变化的能量最小化模拟。
[0076]
然后在步骤s208中,根据所在的高性能计算服务器或超级计算系统来编写相应的作业提交脚本,并且在作业提交后调用系统的分子动力学程序来模拟基态结构。优先地,模拟程序使用lammps(大规模原子分子并行模拟器,large-scale atomic/molecular massively parallel simulator)。该程序由美国sandia国家实验室开发,主要用于分子动力学相关的一些计算和模拟工作。lammps可以支持包括气态、液态或者固态相形态下、各种系综下、百万级的原子分子体系,并提供支持多种势函数。所有计算均使用lammps程序进行,结构文件、势函数参数和输入文件均采用lammps兼容的格式。具体地,结构弛豫使用npt(等压等温)方法进行模拟,采用lammps中的fix npt命令,在温度300k、压强0bar的条件下模拟50万步,然后在温度0.001k、压强0bar的条件下模拟20万步,时间步长为每步1飞秒(femtosecond)。
[0077]
然后在步骤s209中,在作业完成后,从作业的输出信息中获得超胞结构弛豫后的能量值和结构尺寸。
[0078]
然后该方法200以步骤s209中获得的基态结构的性质,对步骤s205中分析出的每个弹性系数进行计算。在步骤s210中判断是否所有弹性系数都进行了计算,若是,则流程进入步骤s214,若否,则进入步骤s211。
[0079]
步骤s211中,对于每个弹性系数的计算,用超胞结构在变形方向上分别作1%、0.5%、-0.5%、-0.1%的拉伸或剪切形变,构造出四个变形子结构;
[0080]
然后步骤s212中,以与步骤s208中相同的方式和设置分别为四个变形子结构提交lammps计算作业进行计算,获得四个变形子结构的弛豫能量值。具体地,可以使用nvt(等温等体)方法对每个变形子结构进行模拟,采用lammps程序中的fix nvt命令,在温度300k的条件下模拟20万步,然后在温度0.001k的条件下模拟20万步,时间步长为每步1飞秒。
[0081]
然后步骤s213中,将四个变形子结构的弛豫能量值与步骤s209中获得的超胞结构
的弛豫能量值以形变为变量拟合出一条能量-体积二次曲线,用所述曲线根据前述公式(1)计算出弹性系数。
[0082]
然后返回步骤s210,重复前述步骤s211至s213,直至所有弹性系数计算完毕。
[0083]
然后该方法200以步骤s209中获得的基态结构的性质,对步骤s205中分析出的每个点缺陷计算点缺陷形成能进行计算。在步骤s214中判断是否所有点缺陷都计算完毕,若是,则流程进入步骤s218,若否,则进入步骤s215。
[0084]
步骤s215中,针对每个点缺陷,用超胞结构构建出相应的点缺陷结构。点缺陷包括空位缺陷和间隙缺陷。对于空位缺陷,应为原胞结构内的每种元素的每种站位分别构建一个空位缺陷结构;对于间隙缺陷,应为原胞结构内的每一处较大空间为每种元素分别构建一个间隙缺陷结构。较大空间可定义为,在结构内部如果能放入一个半径大于的球体且球内不包括任何原子,则球体所在的空间可认为较大空间,间隙缺陷的原子应放在球心位置处。
[0085]
然后步骤s216中,以与步骤s208中相同的方式和设置分别为每个点缺陷结构提交lammps计算作业进行计算,获得每个点缺陷结构的弛豫能量值。具体地,点缺陷结构使用nvt(等温等体)方法进行模拟,采用lammps程序中的fix nvt命令,在温度300k的条件下模拟50万步,然后在温度0.001k的条件下模拟50万步,时间步长为每步1飞秒。
[0086]
然后步骤s217中,将所获得的点缺陷结构的弛豫能量值与步骤s209中获得的超胞结构的弛豫能量值根据前述公式(2)进行计算获得点缺陷形成能。
[0087]
然后返回步骤s214,重复前述步骤s215至s217,直至所有点缺陷计算完毕。
[0088]
然后该方法200以步骤s209中获得的基态结构的性质,对步骤s205中分析出的每个表面结构计算表面形成能。在步骤s218中判断是否所有表面形成能都计算完毕,若是,则流程回到步骤s203,若否,则进入步骤s219。
[0089]
步骤s219中,用超胞结构构建出相应的表面结构。如前所述,原胞结构的表面结构包括常用的小指数界面,miller index指数不超过3,例如(100)、(111)、(310)面。用超胞结构构建出的表面结构的长度、宽度、厚度和真空层均需超过所用势函数的截断距离的两倍。优选地,长度和宽度至少为厚度和真空层至少为
[0090]
然后步骤s220中,以与步骤s208中相同的方式和设置分别为每个表面结构提交lammps计算作业进行计算,获得每个表面结构的弛豫能量值。具体地,表面结构使用nvt(等温等体)方法进行模拟,采用lammps程序中的fix nvt命令,在温度300k的条件下模拟50万步,然后在温度0.001k的条件下模拟50万步,时间步长为每步1飞秒。
[0091]
然后步骤s221中,将所获得的表面结构的弛豫能量值与步骤s209中获得的超胞结构的弛豫能量值根据前述公式(3)进行计算获得表面形成能。
[0092]
然后返回步骤s218,重复前述步骤s219至s221,直至所有表面形成能计算完毕,返回步骤s203。
[0093]
根据本技术上述实施例的基于材料结构模拟的分子动力学势函数的评估方法200可以在最小的人工干预下,在超级计算或高性能计算平台上进行流水线式的高通量运行,实现对势函数的批量快速评估和筛选,大大减轻了科研工作者在进行材料模拟前的前期工作。
[0094]
下面将以铀金属的comb势函数为例,评估其对于α-u和γ-u结构的模拟准确度。
[0095]
步骤301:从用户输入中得到comb势函数文件和参数,以及要模拟的α-u和γ-u结构信息。
[0096]
步骤302:首先对α-u结构进行模拟。α-u结构的基态原胞结构(参见图3所示)所在的空间群为cmcm,三边边长为在考虑了空间对称性后,结构有1个原子站位,坐标为(0,0.10,0.25)。
[0097]
步骤303:α-u结构为正交晶系,有9个弹性系数;有1个原子站位,因此能构建1个空位缺陷,坐标为(0,0.10,0.25);结构中有2处较大的空隙,因此能构建2个间隙缺陷,坐标为(0.5,0.30,0.38)和(0,0.41,0.45)。
[0098]
步骤304:根据步骤302的原胞结构信息构建出一个11
×6×
7的超胞结构,总原子数为1848,三边边长分别为(参见图4所示)。
[0099]
步骤305:从输入的势函数信息按照lammps格式设置输入文件,其中包括但不限于units,pair_style,pair_coeff,thermo_style,timestep,dump,fix,run等参数。
[0100]
units
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
metal
[0101]
pair_style
ꢀꢀꢀꢀꢀꢀꢀ
comb3 polar_off
[0102]
pair_coeff
ꢀꢀꢀꢀꢀꢀ
**u o
[0103]
thermo_style
ꢀꢀꢀꢀꢀ
custom step elapsed temp etotal
[0104]
thermo_modify norm yes
[0105]
timestep
ꢀꢀꢀꢀꢀꢀꢀꢀꢀ
0.001
[0106]
dump
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
datalmp all custom 100000datalmp.*id type q x y z
[0107]
fix
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
1all npt temp 300 300 0.01 tri 0 0 0.1
[0108]
run
ꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀꢀ
500000
[0109]
步骤306:在超算系统中生成作业提交脚本run.sh:
[0110]
#!/bin/bash
[0111]
#sbatch-j job
[0112]
#sbatch-n 1
[0113]
#sbatch-n 32
[0114]
mpirun-np 32lmp-in input》output
[0115]
并使用命令sbatch run.sh来提交作业。
[0116]
步骤307:在作业输出信息中获得结构的总能量为-9798.1ev,原子平均能量为-5.302ev。在作业的最后一个输出结构中,得到三边边长为5.302ev。在作业的最后一个输出结构中,得到三边边长为总体积为则基态的原胞结构的边长为:
[0117][0118][0119][0120]
步骤308:由步骤303分析可知α-u共有c
11
、c
12
、c
13
、c
22
、c
23
、c
33
、c
44
、c
55
、c
66
等9个弹性系数。对于c
11
,把超胞结构向x方向进行拉伸1%、0.5%、-0.5%、-1%,构建出边长为30.82,30.97,31.29,的四个变形子结构;变形子结构在y和z方向的边长均保持在
35.05和使用与步骤305相同的输入设置,以与步骤306相同的方式把这四个变形子结构提交作业进行计算。
[0121]
步骤309:在步骤308计算结束后分别获得弛豫能量值-9795.21、-9797.37、-9797.37、-9795.21ev。
[0122]
步骤310:把步骤309的四个变形子结构和原超胞结构的弛豫能量值一同拟合出一条二次项的能量-体积曲线(参见图5所示),求出拟合公式为e=2.095
×
10-5v2-1.557v 19127,再使用公式(1)计算出
[0123][0124]
步骤311:将超胞结构在y方向拉伸1%、0.5%、-0.5%、-1%,构建四个变形子结构,求出能量-体积曲线,得出c
22
=139gpa。同理,在z、xy、xz、yz等方向上构建变形子结构,得出c
33
、c
12
、c
13
、c
23
。在x面往y和z方向、在y面往z方向构建剪切变形子结构,可得出c
44
、c
55
、c
66
等系数,如下表1。
[0125]
表1:α-u结构的弹性系数(单位:gpa)。
[0126]
c11c12c13c22c232496143139123c33c44c55c66 214411826 [0127]
步骤312:根据步骤303中的结构信息,以步骤307的结构边长构建1个空位缺陷结构(参见图6)和2个间隙缺陷结构(参见图7a和图7b)。使用与步骤305相同的输入设置,以与步骤306相同的方式把这三个点缺陷结构提交作业进行计算。
[0128]
步骤313:在步骤312计算结束后分别获得弛豫能量值-9790.9,-9800.8,-9800.8ev。
[0129]
步骤314:把步骤313中的点缺陷结构的弛豫能量值按公式(2)进行计算。因此,空位缺陷的形成能为:
[0130]
ev=-9790.9-1847(-5.302)=1.9ev
[0131]
间隙缺陷的形成能为:
[0132]ei
=-9800.8-1849(-5.302)=2.6ev
[0133]
步骤315:根据步骤303中的结构信息,以步骤307的结构边长构建出(001)表面结构,长宽为厚度真空层如图8所示。
[0134]
步骤316:使用与步骤305相同的输入设置,以与步骤306相同的方式把步骤315中的表面结构提交作业进行计算,并在作业完成后提取出弛豫能量值-9790.9ev。
[0135]
步骤317:把步骤316中的点缺陷结构的弛豫能量值按公式(3)进行计算。因此,(001)表面的形成能为:
[0136][0137]
步骤318:在计算完α-u结构的性能后,开始计算γ-u结构。γ-u结构的原胞结构
(参见图9所示)的三边边长均为原子坐标为(0,0,0)、(0.5,0.5,0.5)。该结构为立方晶系,有3个弹性系数,能构建出1个空位缺陷,1个间隙缺陷和3种表面结构。
[0138]
步骤319:根据原胞结构的尺寸,构建出一个9
×9×
9的超胞结构,总原子数为1458,边长为(参见图10所示)。
[0139]
步骤320:使用在步骤305设置好的势函数文件以步骤306的方式在超算系统中提交作业。
[0140]
步骤321:在输出信息中获得基态γ-u的总能量为-7718.65ev,原子平均能量为-5.294ev。在输出结构中,得到三边边长为总体积为则原胞结构的实际边长为:
[0141]
步骤322:γ-u结构需要计算c
11
、c
12
、c
44
等3个弹性系数。对于c
11
,把超胞结构向x方向进行拉伸1%、0.5%、-0.5%、-1%,构建出边长为30.44,30.60,30.90,的四个变形子结构,变形子结构在y和z方向的边长均保持在使用与步骤305相同的输入设置,以与步骤306相同的方式把这四个变形子结构提交作业进行计算。
[0142]
步骤323:在步骤322计算结束后分别获得弛豫能量值-7717.13、-7718.27、-7718.27、-7717.13ev。
[0143]
步骤324:把步骤323中的4个变形子结构和原超胞结构的弛豫能量值一同拟合出一条二次项的能量-体积曲线(参见图11所示),求出拟合公式为e=1.795
×
10-5v2-1.0438v 7455.4,再使用公式(1)计算出
[0144][0145]
步骤325:在xy方向上构建拉伸变形子结构,在x面往y方向构建剪切变形子结构,可分别得出c
12
和c
44
系数。
[0146]
表2:γ-u结构的弹性系数(单位:gpa)。
[0147]c11c12c44
16710450
[0148]
步骤326:根据步骤318中的结构信息,以步骤321的结构边长构建1个空位缺陷结构(参见图12所示)和1个间隙缺陷结构(参见图13所示)。使用与步骤305相同的输入设置,以与步骤306相同的方式把这两个点缺陷结构提交作业进行计算。
[0149]
步骤327:在步骤326计算结束后分别获得弛豫能量值-7712.0和-7721.8ev。
[0150]
步骤328:把步骤327中的弛豫能量值代入公式(2),得到空位缺陷形成能:
[0151]
ev=-7712.0-(1458-1)(-5.294)=1.4ev
[0152]
以及间隙缺陷形成能:
[0153]ei
=-7721.8-(1458 1)(-5.294)=2.1ev
[0154]
步骤329:根据步骤318可知γ-u有3种表面结构。构建出(100)表面,长宽为30.75
×
30.75,厚度包含1458个原子(参见图14a所示);构建出(110)表面,长宽为33.83
×
30.75,厚度包含1764个原子(参见图14b所示);构建出(111)表面,长宽为
33.48
×
33.83,厚度为包含1848个原子(参见图14c所示)。所有表面的真空层均为
[0155]
步骤330:使用与步骤305相同的输入设置,以与步骤306相同的方式把步骤329中的表面结构提交作业进行计算,并在作业完成后提取出弛豫能量值-7403.0、-9072.0、-9425.2。
[0156]
步骤331:把步骤330中的弛豫能量值代入公式(3),分别得出(100)表面、(110)表面、(111)表面的表面形成能为:
[0157][0158][0159][0160]
至此所有步骤执行完毕。
[0161]
基于前述实施例的基于材料结构模拟的分子动力学势函数的评估方法,本技术还提出一种系统400。参见图15所示,该系统400包括处理器41和存储器42,处理器41和存储器42通信连接。存储器42存储有计算机程序,所述计算机程序被处理器41执行时使处理器41实现本技术前述实施例的基于材料结构模拟的分子动力学势函数的评估方法。
[0162]
本技术还提出一种计算机可读存储介质,存储有计算机程序,所述计算机程序被处理器执行时实现本技术前述实施例的基于材料结构模拟的分子动力学势函数的评估方法。
[0163]
以上所述仅为本技术的较佳实施例而已,并不用以限制本技术,凡在本技术的精神和原则之内所作的任何修改、等同替换和改进等,均应包含在本技术的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。