1.本发明涉及一种2-烷基环己酮同系物的制备方法,属于香料合成领域。
背景技术:
2.2-烷基环己酮同系物是一种重要的精细化工中间体,可用于制备诸多重要的下游产品。如2-丁基环己酮依次通过baeyer-villiger氧化、皂化和脱水反应可制备得到牛奶内酯,其是一种重要的香料,具有牛奶香味,可用于配制牛奶、奶油、酸奶和草莓等多种类型的香精,应用十分广泛。目前,已见文献报道的2-丁基环己酮合成的方法包括:1)以环己酮为原料,在n-甲基肼作用及三氟乙酸的催化下经过一系列的脱质子化和溴代烷的水解;2)由辛二酸出发,经酯化、缩合、烃化和水解脱羧;3)以正丁醛和环己酮为起始原料,经羟醛缩合和加氢还原。其中,通过正丁醛和环己酮羟醛缩合和选择性加氢制备2-丁基环己酮的工艺实用,原料易得,是最主要的合成方法。以2-丁基环己酮为代表的传统的烷基环己酮合成方法为两步法,首先将醛和环己酮同系物在碱水作用下进行羟醛缩合反应,分离出的缩合产品再进行选择性催化加氢,得到烷基环己酮。
3.如中国发明专利申请公开说明书cn1266841a公开了以正丁醛和环己酮为原料,通过在反应体系中添加氢氧化钠催化羟醛缩合,萃取蒸馏后再添加pd/c催化剂选择性加氢分步反应制备2-丁基环己酮,由于分步反应需要提纯,而氢氧化钠的添加使产品后处理复杂,导致整个合成路线成本高,产生污水多,催化剂难以回收。
4.中国发明专利申请公开说明书cn109651128a和cn109678699a均公开了一种牛奶内酯的连续化生产方法,均包括在碱水作用条件下进行羟醛缩合反应,随后经加氢反应、氧化扩环,最后经酸连续化水解,脱水,得到牛奶内酯香料产品。
5.中国发明专利申请公开说明书cn103864601a公开了以正丁醛和环己酮为原料在相转移催化剂作用下进行碱性缩合,脱水后催化加氢得到2-丁基环己酮的方法,但仍然需要在naoh溶液中反应。
6.然而,这种两步法步骤繁琐,合成路线成本高,产物收率较低,催化剂难以回收,同时还会产生大量的碱性废水,环境污染严重。因此,开发出固体碱作为催化中心,催化羟醛缩合反应有经济价值及环保意义。
7.如中国发明专利cn102019177b公开了一种以复合氧化物为载体,负载至少一种碱性金属氧化物的复合材料进行羟醛缩合,避免了碱性水溶液的使用,减少有机废水的处理排放,且对羟醛缩合反应有优异的催化性能。
8.如中国发明专利cn101205170b公开了使用氧化铝基固体催化剂负载碱金属或碱土金属氧化物以及过渡金属氧化物,催化环己酮自缩合。
9.但由于上述发明中所使用的固体碱的碱强度弱于液体碱,导致羟醛缩合反应速度较慢,因此固体碱催化缩合反应往往需要较高温度获得高反应速率,同时由于随反应的进行原料占比下降、缩合产物比例上升导致缩合反应速率显著下降,副反应增加,选择性下降,为此通过开发烷基环己酮的绿色合成新工艺和新催化剂就显得非常重要。
技术实现要素:
10.针对现有2-烷基环己酮同系物合成方法的不足,本发明提供一种新的2-烷基环己酮同系物的合成方法,实现了2-烷基环己酮同系物的高效、绿色、简单、安全的合成。
11.一种2-烷基环己酮同系物的制备方法,将含有脂肪醛和环己酮和/或环己酮同系物的原料在具有氢气气氛的密闭反应容器中与催化剂接触反应,得到所述2-烷基环己酮同系物;所述催化剂包括多孔碱性氧化物载体、碱性活性组分和加氢活性组分;所述多孔碱性氧化物载体选自氧化锌、氧化钛、氧化锆、氧化铝、na-zsm-5、nay和na-beta分子筛中的至少一种;所述碱性活性组分含有碱性活性元素;所述碱性活性元素选自碱金属元素、碱土金属元素或稀土金属元素中的至少一种;所述加氢活性组分含有加氢活性元素;所述加氢活性元素选自pt元素、pd元素、ru元素、rh元素、ni元素中的至少一种。
12.本发明中,“环己酮同系物”,是指可含αh且在2-6位具有烷基取代基的环己酮。
13.本发明中,“脂肪醛”,是指c
3-c
11
的一元饱和醛,优选为c
3-c8的一元饱和醛。
14.作为优选的技术方案,所述碱金属元素选自钠和/或钾元素;所述碱土金属元素选自钙、镁或钡中的至少一种;所述稀土金属元素选自钇、镧、铈中的至少一种。
15.作为优选的技术方案,所述催化剂中,碱性活性组分含量为0.5%~10%,加氢活性组分含量为0.05%~10%;其中,所述碱性活性组分的含量以其中所含的碱性活性元素的质量百分数计;所述加氢活性组分的含量以其中所含加氢活性元素的质量百分数计;
16.优选地,加氢活性组分含量为0.1%~2%,碱性活性组分含量为0.5%~5%;
17.优选地,所述加氢活性组分含量的上限选自0.1%、0.2%、0.5%、1%、1.5%、2%、2.5%、5%、7%、9%或10%;下限选自0.05%、0.1%、0.2%、0.5%、1%、1.5%、2%、2.5%、5%、7%或9%;
18.优选地,所述碱性活性组分含量的上限选自1%、1.5%、2%、3%、5%、6%、7%、8%、9%或10%;下限选自0.5%、1%、1.5%、2%、3%、5%、6%、7%、8%或9%。
19.作为优选的技术方案,所述环己酮和/或环己酮同系物与脂肪醛的摩尔比为1:1~10:1;
20.优选地,环己酮和/或环己酮同系物与脂肪醛的摩尔比为2:1~5:1;
21.优选地,所述含α-h的环己酮同系物环己酮与醛类的摩尔比的上限选自2:1、3:1、4:1、5:1、6:1、7:1、8:1、9:1或10:1;下限选自1:1、2:1、3:1、4:1、5:1、6:1、7:1、8:1或9:1。
22.作为优选的技术方案,所述催化剂的用量为原料总质量的0.5%~15%;
23.优选地,所述催化剂的用量为原料总质量的0.5%~10%;
24.优选地,所述催化剂的用量为原料总质量的上限选自0.8%、0.83%、1%、2%、3%、5%、6%、7%、8%、9%、10%或15%;下限选自0.5%、0.8%、0.9%、1%、2%、3%、5%、6%、7%、8%、9%或10%。
25.作为优选的技术方案,所述环己酮同系物为含羰基α氢的环己酮同系物;
26.优选地,所述环己酮同系物选自环己酮、2-乙基环己酮、3-丙基环己酮、4-异丙基环己酮或4-甲基环己酮中的至少一种;
27.优选地,所述脂肪醛选自丙醛、正丁醛、正戊醛、正己醛、正庚醛、正辛醛、异丁醛、正异戊醛中的至少一种。
28.作为优选的技术方案,所述反应容器中氢气的压力为0.5~5.0mpa;
29.优选地,所述反应容器中氢气的压力为1.0~4.0mpa。
30.优选地,所述氢气的压力为0.5~4.5mpa。
31.优选地,所述氢气的压力为1.0~3.5mpa。
32.优选地,所述氢气的压力为0.5~4mpa。
33.优选地,所述氢气的压力为1.0~3.0mpa。
34.优选地,所述氢气的压力的上限选自1mpa、1.5mpa、2mpa、2.5mpa、3mpa、3.5mpa、4mpa、4.5mpa或5mpa;下限选自0.5mpa、1mpa、1.5mpa、2mpa、2.5mpa、3mpa、3.5mpa、4mpa或4.5mpa。
35.作为优选的技术方案,所述反应的温度为80~180℃;
36.优选地,所述反应的温度为100~160℃。
37.优选地,所述反应的温度的上限选自110℃、120℃、130℃、140℃、150℃、160℃、170℃或180℃;下限选自100℃、110℃、120℃、130℃、140℃、150℃、160℃或170℃。
38.优选地,所述密闭反应容器的氢气的压力不再降低时,反应终止。
39.本发明中,实际反应中随着氢气的消耗,氢气的压力会缓慢下降,溶剂在催化剂的作用下会发生自缩合加氢,实验室中也可通过在线取样得到釜中样品来确定反应程度。
40.本发明的另一个方面,提供一种用于制备2-烷基环己酮同系物的催化剂的制备方法,至少包括以下步骤:将多孔碱性载体或其前驱体浸渍于含有碱性活性元素和加氢活性元素的溶液后,干燥,煅烧,得到所述催化剂,其中,可溶性盐优选为对应乙酸盐、草酸盐、硝酸盐或氯化物中的至少一种。
41.本发明的催化剂的制备方法可以通过常规的浸渍法合成,也可以用共沉淀的方法来负载贵金属(如:孙春晖,于海斌,陈永生,许岩,刘伟.共沉淀法制备ni/al2o3催化剂的研究[j].无机盐工业,2014,46(11):76-78.共沉淀法制备ni/al2o3催化剂的研究)。
[0042]
作为一种具体的实施方式,2-烷基环己酮同系物的制备方法至少包括:
[0043]
将含有脂肪醛、含αh的环己酮同系物和催化剂的混合物混合均匀,在反应温度80~180℃和氢气压力0.5~5.0mpa条件下进行羟醛缩合和加氢反应,生成2-烷基环己酮同系物。
[0044]
可选地,所述反应无需任何溶剂。
[0045]
本发明能产生的有益效果包括:
[0046]
本发明提供的2-烷基环己酮同系物的制备方法,催化脂肪醛和含α-h的环己酮同系物一步法制备2-烷基环己酮同系物,通过在催化剂中加入碱金属、碱土金属或稀土金属元素助剂,同时通过耦合缩合产物的加氢反应移除缩合产物,使得2-烷基环己酮同系物的制备具有原料转化率高,选择性好,催化活性稳定和绿色环保的优点,操作压力低,且在反应过程中,无需调整反应温度和压力,原料脂肪醛类转化率大于90%,2-烷基环己酮同系物选择性大于80%,可实现2-烷基环己酮同系物的高效、绿色、简单、安全的合成,操作工艺简单,具有良好的应用前景。
附图说明
[0047]
图1为实施例21中cat-29在相同条件下重复使用10次的转化率和选择性柱状图。
[0048]
图2为对实施例中产物精馏所得2-丁基环己酮的核磁共振碳谱。
[0049]
图3为对实施例中产物精馏所得2-丁基环己酮的核磁共振氢谱。
[0050]
图4为对比例3中cat-40在相同条件下重复使用10次的转化率和选择性柱状图。
具体实施方式
[0051]
下面结合实施例详述本技术,但本发明并不局限于这些实施例。
[0052]
如无特别说明,本发明的实施例中的原料均通过商业途径购买。实施例中,如无特别说明,所用技术手段为本领域常规的技术手段。实施例中,如无特别说明,所给仪器的测试方法均采用厂家推荐的设置。
[0053]
本发明的实施例中分析方法如下:
[0054]
反应后的产物组成在agilent 7890气相色谱上进行分析,采用db-5色谱柱和fid检测器,通过峰面积归一化法计算产物的选择性,转化率基于脂肪醛的消耗来计算。
[0055]
本发明的实施例中转化率、选择性计算如下,转化率以及选择性都基于脂肪醛的摩尔数进行计算。
[0056][0057][0058]
i为脂肪醛转化产物,除目标产物2-烷基环己酮同系物外,还包括如脂肪醇、脂肪醛自缩合产物、烯烃及其多聚体等副产物。
[0059]
实施例1催化剂制备
[0060]
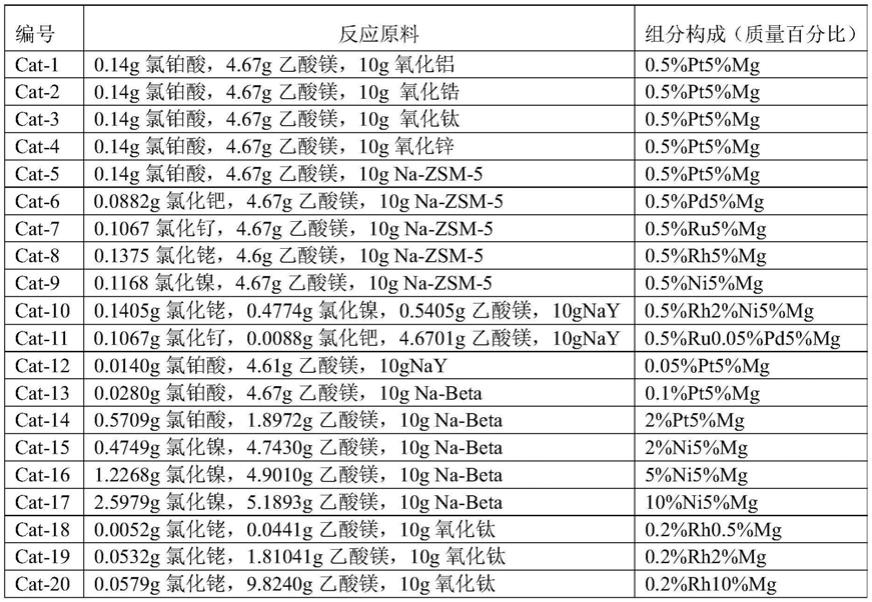
将一定量的铂、钯、钌、铑和镍活性组分中的一种或两种组合对应的可溶性盐与一定量的钾、钠、镁、钙、钡、钇、镧和铈助剂中的一种或两种组合对应的可溶性盐溶解于去离子水中,形成均匀的浸渍溶液,将氧化锌、氧化钛、氧化锆、氧化铝或钠型分子筛等碱性固体多孔氧化物分别浸渍于上述浸渍溶液中,浸渍一定时间后,于80~120℃烘箱中将水分蒸发,所得固体前驱体于450℃焙烧,得到目标催化剂,共制得34个催化剂,记为cat-1~cat-34,催化剂反应原料和组分构成列于表1。
[0061]
表1实施例1中所得催化剂的组成
[0062][0063][0064]
实施例2 2-丁基环己酮的制备
[0065]
分别称取2.46g实施例1中得到的cat-1~cat-34催化剂加入到有36g环己酮和13.2g正丁醛的反应釜中,将反应釜密封后,通入氮气使反应釜气压达到0.2mpa,通过排气口将反应釜的气体排出,重复三次,再通入氢气1mpa,再排气重复三次,再充入2.5mpa的氢气,开启搅拌充分混合后,升温至140℃,恒温直至釜内反应压力不再降低,结束反应,冷却,排气,过滤产物,液体产物依次记为pro-1~pro34。
[0066]
转化率和产物的选择性,结果列于表2。
[0067]
实施例3 2-丁基环己酮的制备
[0068]
分别称取0.41g,2.95g和4.92g实施例1中得到的cat-6催化剂加入到有36g环己酮
和13.2g正丁醛的反应釜中,其余操作与实施例2中完全相同,所得产物依次记为pro-35~pro-37,转化率和产物的选择性,结果列于表2。
[0069]
实施例4 2-丁基环己酮的制备
[0070]
称取0.94g实施例1中得到的cat-6催化剂加入到有18g环己酮和13.2g正丁醛的反应釜中,其余操作与实施例2中完全相同,所得产物记为pro-38。
[0071]
实施例5 2-丁基环己酮的制备
[0072]
称取3.1g实施例1中得到的cat-6催化剂加入到有90g环己酮和13.2g正丁醛的反应釜中,其余操作与实施例2中完全相同,所得产物记为pro-39。
[0073]
实施例6 2-丁基环己酮的制备
[0074]
称取5.8g实施例1中得到的cat-6催化剂加入到有180g环己酮和13.2g正丁醛的反应釜中,其余操作与实施例2中完全相同,所得产物记为pro-40。
[0075]
实施例7 2-丁基环己酮的制备
[0076]
称取2.46g实施例1中得到的cat-6催化剂加入到有36g环己酮和13.2g正丁醛的反应釜中,反应温度为100℃,其余操作与实施例2中完全相同,所得产物记为pro-41。
[0077]
实施例8 2-丁基环己酮的制备
[0078]
称取2.46g实施例1中得到的cat-6催化剂加入到有36g环己酮和13.2g正丁醛的反应釜中,反应温度为160℃,其余操作与实施例2中完全相同,所得产物记为pro-42。
[0079]
实施例9 2-丁基环己酮的制备
[0080]
称取2.46g实施例1中得到的cat-6催化剂加入到有36g环己酮和13.2g正丁醛的反应釜中,反应温度为180℃,其余操作与实施例2中完全相同,所得产物记为pro-43。
[0081]
实施例10 2-丁基环己酮的制备
[0082]
称取2.46g实施例1中得到的cat-6催化剂加入到有36g环己酮和13.2g正丁醛的反应釜中,反应压力为0.5mpa,其余操作与实施例7中完全相同,所得产物记为pro-44。
[0083]
实施例11 2-丁基环己酮的制备
[0084]
称取2.46g实施例1中得到的cat-6催化剂加入到有36g环己酮和13.2g正丁醛的反应釜中,反应压力为1.0mpa,其余操作与实施例7中完全相同,所得产物记为pro-45。
[0085]
实施例12 2-丁基环己酮的制备2-丁基环己酮的制备
[0086]
称取2.46g实施例1中得到的cat-6催化剂加入到有36g环己酮和13.2g正丁醛的反应釜中,反应压力为4.5mpa,其余操作与实施例7中完全相同,所得产物记为pro-46。
[0087]
实施例13 2-丙基环己酮的制备
[0088]
称取实施例1中得到的cat-4催化剂2.46g加入到有36g环己酮和13.2g正丙醛的反应釜中,其余操作与实施例2中完全相同,所得产物依次记为pro-47。
[0089]
实施例14 2-戊基环己酮的制备
[0090]
称取实施例1中得到的cat-4催化剂2.46g加入到有36g环己酮和13.2g正戊醛的反应釜中,其余操作与实施例2中完全相同,所得产物依次记为pro-48。
[0091]
实施例15 2-己基环己酮的制备
[0092]
称取实施例1中得到的cat-4催化剂2.46g加入到有36g环己酮和13.2g正己醛的反应釜中,其余操作与实施例2中完全相同,所得产物依次记为pro-49。
[0093]
实施例16 2-庚基环己酮的制备
[0094]
称取实施例1中得到的cat-4催化剂2.46g加入到有36g环己酮和13.2g正庚醛的反应釜中,其余操作与实施例2中完全相同,所得产物依次记为pro-50。
[0095]
实施例17 2-辛基环己酮的制备
[0096]
称取实施例1中得到的cat-4催化剂2.46g加入到有36g环己酮和13.2g正辛醛的反应釜中,其余操作与实施例2中完全相同,所得产物依次记为pro-51。
[0097]
实施例18 2-异丁基环己酮的制备
[0098]
称取实施例1中得到的cat-4催化剂2.46g加入到有36g环己酮和13.2g异丁醛的反应釜中,其余操作与实施例2中完全相同,所得产物依次记为pro-52。
[0099]
实施例19 2-丁基,6-乙基环己酮的制备
[0100]
称取实施例1中得到的cat-4催化剂2.46g加入到有36g2-乙基环己酮和13.2g正丁醛的反应釜中,其余操作与实施例2中完全相同,所得产物依次记为pro-53。
[0101]
实施例20 2-丁基,4-异丙基环己酮的制备
[0102]
称取实施例1中得到的cat-4催化剂2.46g加入到有36g4-异丙基环己酮和13.2g正丁醛的反应釜中,其余操作与实施例2中完全相同,所得产物依次记为pro-54。
[0103]
实施例21 cat-29稳定重复性实验
[0104]
称取2.46g实施例1中cat-29催化剂加入到有36g环己酮和13.2g正丁醛的反应釜中,将反应釜密封后,通入氮气使反应釜气压达到0.2mpa,通过排气口将反应釜的气体排出,重复三次,再通入氢气1mpa,再排气重复三次,再充入2.5mpa的氢气,开启搅拌充分混合后,升温至140℃,恒温直至釜内反应压力不再降低,结束反应。
[0105]
冷却,排气,静止催化剂完全沉降后,移走液体,加入36g环己酮和13.2g正丁醛,重复以上操作,共进行10次实验。
[0106]
所得产物分析以此记为re-1~re-10,数据记录在表3和图1。
[0107]
图1显示了本发明所制备的催化剂不仅具有优良的单程反应性能,同时还具有较稳定的循环使用性能。
[0108]
图2和图3中的核磁共振谱图显示出与目标产物对应的核磁谱图,且未显示碳碳双键以及羟基所对应的化学位移值的峰,说明本专利对制备2-烷基环己酮同系物,如2-丁基环己酮有优良的反应性能。
[0109]
对比例1无碱性活性组分
[0110]
将一定量的铂、钯、钌、铑和镍活性组分中的一种或两种组合对应的可溶性盐溶解于去离子水中,形成均匀的浸渍溶液,将氧化锌、氧化钛、氧化锆、氧化铝或钠型分子筛等碱性固体多孔氧化物分别浸渍于上述浸渍溶液中,浸渍一定时间后,于80~120℃烘箱中将水分蒸发,所得固体前驱体于450℃焙烧,得到目标催化剂,共制得5个催化剂,记为cat-35~cat-40,催化剂反应原料和组分构成列于表2。
[0111]
表2对比例1中所得催化剂的组成
[0112]
催化剂编号反应原料组分构成(质量百分比)cat-350.1334g氯铂酸,10g氧化锌0.5%ptcat-360.1334g氯铂酸,10g氧化钛0.5%ptcat-370.1334g氯铂酸,10g氧化锆0.5%ptcat-380.1334g氯铂酸,10g氧化铝0.5%pt
cat-390.1334g氯铂酸,10g na-zsm-50.5%ptcat-400.0882g氯化钯,10g氧化铝0.5%pd
[0113]
对比例2 2-丁基环己酮的制备
[0114]
分别称取2.46g对比例1中得到的cat-35~cat-39催化剂加入到有36g环己酮和13.2g正丁醛的反应釜中,将反应釜密封后,通入氮气使反应釜气压达到0.2mpa,通过排气口将反应釜的气体排出,重复三次,再通入氢气1mpa,再排气重复三次,再充入2.5mpa的氢气,开启搅拌充分混合后,升温至140℃,恒温直至釜内反应压力不再降低,结束反应,冷却,排气,过滤产物,产物依次记为pro-55~pro59。
[0115]
对比例3 cat-40稳定重复性实验
[0116]
称取2.46g对比例1中cat-40催化剂加入到有36g环己酮和13.2g正丁醛的反应釜中,将反应釜密封后,通入氮气使反应釜气压达到0.2mpa,通过排气口将反应釜的气体排出,重复三次,再通入氢气1mpa,再排气重复三次,再充入2.5mpa的氢气,开启搅拌充分混合后,升温至140℃,恒温直至釜内反应压力不再降低,结束反应。
[0117]
冷却,排气,静止催化剂完全沉降后,移走液体,加入36g环己酮和13.2g正丁醛,重复以上操作,共进行10次实验。
[0118]
所得产物分析以此记为are-1~are-10,数据记录在表3和图4。
[0119]
图1和图4对说明了本发明所制备的催化剂的稳定性明显高于单一氧氧化物如化铝载体催化剂。推测原因是单一氧化物如氧化铝的水热稳定性一般较差,有文献表明水的存在会影响催化剂的活性和稳定性,而本反应是一个密封式的产水反应,因此催化剂易受到水的影响(如:tai,j.and r.j.davis(2007)."synthesis of methacrylic acid by aldol condensation of propionic acid with formaldehyde over acid
–
base bifunctional catalysts."catalysis today123(1-4):42-49)。
[0120]
对2-烷基环己酮同系物的制备的产物分析结果总结如下表3。
[0121]
表3反应产物分析结果
[0122]
[0123]
[0124][0125]
由表3中数据可知,本发明提供的2-烷基环己酮同系物的合成方法,具有原料转化率高,产物选择性好和绿色环保的优点,且在反应过程中,无需调整反应温度和压力,操作简单安全,具有良好的应用前景。对比着未添加助剂的催化剂cat-55至cat-59,使用助剂的催化剂的醛类转化率在90%以上、产物选择性大于80%,远高于未添加助剂的催化剂的醛类转化率和产物选择性。
[0126]
本发明催化剂具有固体碱和加氢金属双活性中心,可催化脂肪醛和含羰基α-h的环己酮经羟醛缩合和加氢一步法制备2-烷基环己酮同系物。
[0127]
以上所述,仅是本技术的几个实施例,并非对本技术做任何形式的限制,虽然本技术以较佳实施例揭示如上,然而并非用以限制本技术,任何熟悉本专业的技术人员,在不脱
离本技术技术方案的范围内,利用上述揭示的技术内容做出些许的变动或修饰均等同于等效实施案例,均属于技术方案范围内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。