1.本发明属于计算机科学的方法,涉及人工智能对生物信息数据的处理和分析,特别涉及一种提高单细胞深度聚类精度的方法。
背景技术:
2.随着高通量测序技术的发展,我们可以测得单细胞的测序数据,所测得的单细胞数据具有高维度和样本量非常大的特点,因此,传统对基因表达的研究方法不再适用。传统的技术从生物组织层面出发进行数据分析,包括线性地降维和聚类,以确定细胞信息,这种技术无法从单个细胞的层面来研究细胞的异质性。目前一些方法也开始针对单细胞的数据特点进行改进,但仍然以矩阵分解为主,准确性不好,影响下游分析。研究表明,使用深度神经网络可以较好地来处理和分析单细胞数据。现有的深度生成模型主要是以vae(变分自编码器)为基础框架。
3.scvi和scale都是基于深度神经网络分析单细胞的框架方法。scvi是基于变分自编码器的单细胞处理方法,scale是基于变分自编码器和高斯混合模型的框架,也是和本发明最相似的,它使用单细胞表观组数据,具有较高的聚类准确度。scale在vae框架下由编码器和解码器组成。编码器是一个四层神经网络(3200-1600-800-400),解码器是一个只有一层的网络,10维潜变量(特征)直接连接到输出端。潜变量分布在μc和σc参数化的gmm流形上。
4.但是,现有技术存在一些缺陷,主要是准确度不够高,以及需要人为设置细胞类型个数等问题。准确度低,说明了现有方法不一定适用于大规模的单细胞数据,可能无法准确捕捉到数据的有效特征等。而人为地设置聚类的个数,则有可能会影响到下游分析,即人为设置一些参数并不一定是可靠的。
技术实现要素:
5.为了解决上述问题,本发明提供了一种提高单细胞深度聚类算法精度的方法,其特征在于:包括以下步骤:
6.(1)数据处理
7.包括异常细胞和异常基因表达的过滤以及归一化,并进行tf-idf转换;
8.(2)将步骤(1)处理后的数据输入变分自编码器,对步骤(1)得到的数据进行训练
9.所述变分自编码器包括两个神经网络:编码器神经网络和解码器神经网络,以及两个变量:观测变量x和潜变量z;
10.变分自编码器的公式如下:
11.表示变分自编码器在参数λ和参数设置好的情况下输出的x是多少;代表了重构误差,其中q
λ
(z|x)表示编码器的输出,表示解码器的输出,x表示观测变量,z表示隐藏变量,λ和分别表示编码器神经网络和解码器神经网络
的训练参数;kl(q
λ
(z|x)||p(z))代表了近似后验分布和先验分布之间的kl散度,其中q
λ
(z|x)代表近似后验分布,其中x表示观测变量,z表示隐藏变量,λ表示编码器神经网络的训练参数,p(z)表示先验分布,其中z~n(0,1);然后结合变分自编码器和贝叶斯高斯混合模型,推断出聚类的个数,贝叶斯高斯混合模型是高斯混合模型的拓展,接着根据方程表示高斯混合模型是m个分量密度的加权和,其中x是一个d维向量,wi,i=1,...,m表示权重,g(x|μi,σi),i=1,...m表示高斯分量密度;
12.本步骤利用了具有狄利克雷分布的有限混合模型和一个具有狄利克雷过程的无限混合模型,在狄利克雷过程混合模型中,dp被用作层次贝叶斯规范中的非参数先验:
13.g|{α,g0}~dp(α,g0),
14.ηn|g~g,
15.xn|ηn~p(xn|η
ns
),
16.其中,g0表示基本测度,α表示基本测度的离散程度,g表示从基本测度中采样出来的分布,ηn|g是服从g的分布,其中ηn显示出聚类效应,xn表示产生的数据,其可以根据参数的不同值进行区分;
17.由该模型生成的数据可以根据不同的参数值进行分区;
18.dp混合是一种灵活的混合模型,其中组分的数量(即分区中的细胞数量)是随机的,并随着新数据的观察而扩展,利用kolmogorov一致性定理,通过式上中的有限维分布来定义dp;将dp更具体地描述为一种断棍结构,两个随机变量的无限集合,公式如下:
19.vi~beta(1,α),i={1,2,...},
20.其中,vi是由beta(1,α)分布中独立采样得出的随机变量,采样结果决定了每一部分的大小,且与棍子的剩余部分成比例;
[0021][0022]
其中,表示高斯分布的参数
[0023][0024]
其中,是断棍表达式的形式,π(vi)是其对应的采样的权重,
vj
代表前一次采样的权重,v={v1,v2,...,v
∞
}表示无限次的采样的权重集合)
[0025]
p(di|v)=mult(π(v)),
[0026]
其中,mult(π(v))表示以π(v)为参数的多项式分布,p(di|v)表示从多项式分布中采样得到的概率
[0027]
p(d)=p(di|v),
[0028]
其中,把多项式分布p(di|v)表示成p(d),其中d是一个分类变量,其概率是离散的
[0029][0030]
联合概率如下:
[0031]
p(x,z,d)=p(x|z)p(z|d)p(d),
[0032]
训练变分自编码器以达到最大化观测数据的似然函数:
[0033][0034]
它被转换为最大化证据的下界(elbo):
[0035]
elbo(x)=e
q(z,d|x)
[logp(x|z)]-d
kl
(q(z,d|x)||p(z,d)),
[0036]
其中,d
kl
是kl散度值,表示两个分布之间的差距;
[0037]
g|{α,g0}~dp(α,g0),
[0038]
ηn|g~g,
[0039]
xn|ηn~p(xn|η
ns
),
[0040]
g0表示基本测度,α表示基本测度的离散程度,g表示从基本测度中采样出来的分布,ηn|g是服从g的分布,其中ηn显示出聚类效应,xn表示产生的数据,其可以根据参数的不同值进行区分;
[0041]
正则化项是一个kl散度,它将潜变量z正则化为贝叶斯高斯混合模型流形,q(z,c|x)和p(x|z)分别是编码器和解码器,分别用两个神经网络建模,将步骤(1)处理后(包括异常细胞和异常基因表达的过滤以及归一化,并进行tf-idf转换)的数据输入编码器神经网络,对数据进行降维;
[0042]
(3)将步骤(2)降维后的数据进行第一次聚类分析,本步骤采用k-means聚类算法,直接使用从模型推断出来的聚类个数k;
[0043]
(4)进行第二次聚类
[0044]
第二次聚类基于第一次聚类预测的伪标签,采用leiden聚类算法,本步骤中,对第二次聚类的预测结果和第一次聚类得到的伪标签进行评分,即ari:
[0045][0046]
其中,ari是调整兰德系数,ri是兰德系数,expected(ri)是兰德系数的期望值,max(ri)表示兰德系数的最大值,兰德系数ri表达式为:n表示实例总数,用c表示实际的类别划分,k表示聚类结果。定义a为在c中被划分为同一类,在k中被划分为同一簇的实例对数量。定义b为在c中被划分为不同类别,在k中被划分为不同簇的实例对数量,显然ri的值越大说明聚类效果越好;
[0047]
两个簇的结果通过ari评分进行比较,设置γ的取值范围作为比较范围,得分高的结果作为γ参数选择的依据,这样可以从复杂网络的角度对聚类模型进行约束,进一步提高其性能。
[0048]
优选的,所述步骤(1)中数据过滤方式为:对scatac-seq计数矩阵进行了筛选,只保留了至少100个细胞和至少0.01倍于表达细胞数的基因,然后,计算差异性分数,所有细胞差异性特征进行排名,在排名结束后,选择排名前30000的基因进行分析;然后对原始的scatac-seq计数矩阵进行tf-idf转换。
[0049]
优选的,所述步骤(2)中编码器神经网络设置成6层的深度神经网络,维数分别是数据本身维度,3000维,1500维,840维,420维和潜在空间的固定维度10维,再通过解码器神经网络将数据维度还原到其自身的维度,其中的激活函数主要使用了relu函数,批次大小设置为30,优化器采用了adam优化器。
[0050]
优选的,所述步骤(3)第一次聚类分析时,时间复杂度:o(tknm),其中,t为迭代次数,k为簇的数目,n为样本点数,m为样本点维度;空间复杂度:o(m(n k)),其中,k为簇的数目,m为样本点维度,n为样本点数。
[0051]
与现有技术相比,本发明的有益之处为:
[0052]
1.本发明相对于以往的方法(scale,scvi和cicero等方法)提高了聚类的准确性;
[0053]
2.本发明不需要人为地设置聚类个数,这样就避免了人为设置的聚类个数与数据本身细胞类型个数不符所造成的偏差,避免了对下游分析的影响;
[0054]
3.本发明具有一个二次聚类功能,可以在kmeans聚类的初次聚类结果上进一步聚类,相对于自身可以进一步提升聚类的准确性。
附图说明
[0055]
下面结合附图对本发明作进一步的说明。
[0056]
图1是本发明单细胞数据处理和分析的框架流程图;
[0057]
图2是本发明与其他方法的结果对比图,其中,scvabgme是本发明的方法,scale,scvi,cicero是其他另外三种方法,forebrain,insilico,splenocyte,leukemia,gm12878/hl-60,gm12878/hek293t分别是6个sc-atac的单细胞数据集;
[0058]
图3是本发明二次聚类后与其他方法的结果对比图,其中scvabgme是本发明的方法,further clustering是进一步聚类,scale,scvi,cicero是其他另外三种方法,forebrain,insilico,splenocyte,leukemia,gm12878/hl-60,gm12878/hek293t分别是6个sc-atac的单细胞数据集;
[0059]
图4是本发明在forebrain,insilico,splenocyte,leukemia,gm12878/hl-60,gm12878/hek293t 6个单细胞数据集上的聚类结果图,图的右侧分别显示出了识别到的细胞类型。
具体实施方式
[0060]
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述。显然,所描述的实施例仅仅是本发明的一部分实施例,而不是全部的实施例。因此,以下对在附图中提供的本发明的实施例的详细描述并非旨在限制要求保护的本发明的范围,而是仅仅表示本发明的选定实施例。基于本发明的实施例,本领域技术人员在没有做出创造性劳动的前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0061]
实施案例1
[0062]
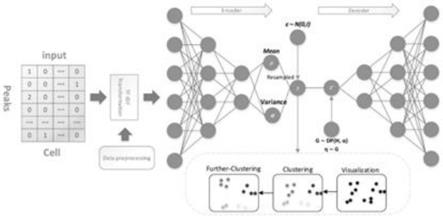
如图1所示,一种提高单细胞深度聚类算法精度的方法,其特征在于:包括以下步骤:
[0063]
(1)数据处理
[0064]
包括异常细胞和异常基因表达的过滤以及归一化,并进行tf-idf转换;本发明对scatac-seq计数矩阵进行了筛选,只保留了至少100个细胞和至少0.01倍于表达细胞数的基因。然后,计算差异性分数,所有细胞差异性特征进行排名,在排名结束后,选择了排名前30000的基因进行分析。然后对原始的scatac-seq计数矩阵进行tf-idf转换。
[0065]
(2)将步骤(1)处理后的数据输入变分自编码器,对步骤(1)得到的数据进行训练
[0066]
变分自动编码器(vae)是一种典型的深度生成模型,它通过低维潜在特征来捕捉高维数据的分布。该模型包括两个神经网络:编码器神经网络和解码器神经网络,以及两个变量:观测变量x和潜变量z;
[0067]
变分自编码器的公式如下:
[0068][0069]
等式左边表示变分自编码器在参数λ和参数设置好的情况下输出的x是多少,等式右边第一项代表了重构误差其中q
λ
(z|x)表示编码器的输出,表示解码器的输出,x表示观测变量,z表示隐藏变量,λ和分别表示编码器神经网络和解码器神经网络的训练参数;第二项kl(q
λ
(z|x)||p(z))代表了近似后验分布q
λ
(z|x)和先验分布p(z)之间的kl散度,q
λ
(z|x)代表近似后验分布,其中x表示观测变量,z表示隐藏变量,λ表示编码器神经网络的训练参数,p(z)表示先验分布,其中z~n(0,1);然后结合变分自编码器和贝叶斯高斯混合模型,推断出聚类的个数,贝叶斯高斯混合模型是高斯混合模型的拓展,高斯混合模型(gmm)是描述一个参数可能性密度函数的高斯分量密度加权和,在单细胞数据的处理中,gmm参数由训练数据通过期望最大化(em)迭代过程估计。根据方程,高斯混合模型是m分量高斯密度的加权和,其中x是一个d维向量,wi,i=1,...,m表示权重,g(x|μi,σi),i=1,...m表示高斯分量密度;贝叶斯高斯混合模型是一种扩展了高斯混合模型的变分推理算法。本发明利用了具有狄利克雷分布的有限混合模型和一个具有狄利克雷过程的无限混合模型,狄利克雷过程推理算法在实践中是近似的,它采用了一个截断分布,具有一个集合的最大数量的组件(称为断棍表示)。实际使用的组件数量几乎总是由数据决定的。这意味着我们不需要去刻意设定集群的数量。在狄利克雷过程混合模型中,dp被用作层次贝叶斯规范中的非参数先验:
[0070]
g|{α,g0}~dp(α,g0),
[0071]
ηn|g~g,
[0072]
xn|ηn~p(xn|η
ns
),
[0073]
其中,g0表示基本测度,α表示基本测度的离散程度,g表示从基本测度中采样出来的分布,ηn|g是服从g的分布,其中ηn显示出聚类效应,xn表示产生的数据,其可以根据参数的不同值进行区分;
[0074]
由该模型生成的数据可以根据不同的参数值进行分区;根据这一解释,dp混合是一种灵活的混合模型,其中组分的数量(即分区中的细胞数量)是随机的,并随着新数据的观察而扩展,利用kolmogorov一致性定理,通过式上中的有限维分布来定义dp;将dp更具体地描述为一种断棍结构,两个随机变量的无限集合,公式如下:
[0075]
vi~beta(1,α),i={1,2,...},
[0076]
vi是由beta(1,α)分布中独立采样得出的随机变量,采样结果决定了每一部分的大小,且与棍子的剩余部分成比例;
[0077][0078]
[0079]
其中,是断棍表达式的形式,π(vi)是其对应的采样的权重,
vj
代表前一次采样的权重,v={v1,v2,...,v
∞
}表示无限次的采样的权重集合
[0080]
p(di|v)=mult(π(v)),
[0081]
其中,mult(π(v))表示以π(v)为参数的多项式分布,p(di|v)表示从多项式分布中采样得到的概率
[0082]
p(d)=p(di|v),
[0083]
其中,把多项式分布p(di|v)表示成p(d),其中d是一个分类变量,其概率是离散的
[0084][0085]
联合概率如下:
[0086]
p(x
,
z,d)=p(x|z)p(z|d)p(d),
[0087]
训练变分自编码器以达到最大化观测数据的似然函数:
[0088][0089]
它被转换为最大化证据的下界(elbo):
[0090]
elbo(x)=e
q(z,d|x)
[logp(x|z)]-d
kl
(q(z,d|x)||p(z,d)),
[0091]
其中,
dkl
是kl散度值,表示两个分布之间的差距
[0092]
g|{α,g0}~dp(α,g0),
[0093]
ηn|g~g,
[0094]
xn|ηn~p(xn|η
ns
),
[0095]
重构项鼓励输入数据与输出数据相似,重构项的目的就是使得输入项与输出项的数据的分布尽可能地相似,也就是使其差距尽可能的小。正则化项是一个kl散度,它将潜变量z正则化为贝叶斯高斯混合模型流形,q(z,c|x)和p(x|z)分别是编码器和解码器,分别用两个神经网络建模,将步骤(1)处理后(包括异常细胞和异常基因表达的过滤以及归一化,并进行tf-idf转换)的数据输入编码器神经网络,对数据进行降维;将编码器神经网络设置成6层的深度神经网络,解码器设置成2层的神经网络,维数分别是数据本身维度,3000维,1500维,840维,420维和潜在空间的固定维度10维,再通过解码器神经网络将数据维度还原到其自身的维度,其中的激活函数主要使用了relu函数,批次大小设置为30,优化器采用了adam优化器,adam优化器是对sgd优化器的扩展,可以代替经典的随机梯度下降法来更有效地更新网络权重。其具有以下优点:计算效率高;较少的内存需求;适用于非常嘈杂和高稀疏梯度的问题,adam使用动量和自适应学习率来加快收敛速度。(3)将步骤(2)降维后的数据进行第一次聚类分析,本步骤采用k-means聚类算法,这是一种基础聚类算法,需要提前设置聚类个数k,其中的聚类个数k不需要像scale(scale method for single-cell atac-seq analysis via latent feature extraction.nature communications,10,1-10)方法中的那样手动设置,这里k直接使用从模型推断出来的聚类个数,伪代码如下:
[0096]
获取数据n个m维的数据
[0097]
随机生成k个m维的点
[0098][0099]
时间复杂度:o(tknm),其中,t为迭代次数,k为簇的数目,n为样本点数,m为样本点维度。
[0100]
空间复杂度:o(m (n k)),其中,k为簇的数目,m为样本点维度,n为样本点数。
[0101]
如图2所示,聚类结束后,使用三种指标对其聚类结果进行评价,分别是:ari、nmi和f1-score,ari是ri的改进,值越大意味着聚类结果与真实情况越吻合。nmi是归一化互信息,f1-score是精确率和召回率的调和平均数,最大为1,最小为0。分别与scale,scvi和cicero三种方法在forebrain,insilico、splenocyte,leukemia,gm12878/hl-60,gm12878/hek293t这6个sc-atac单细胞数据集上进行比较,最终本发明方法的准确率优于现有方法。
[0102]
(4)进行第二次聚类
[0103]
第二次聚类基于第一次聚类预测的伪标签,采用leiden聚类算法,这是一种基于模块化测量和社区发现的聚类方法。但是有一个问题需要解决,即如何在第二次聚类中的γ参数。这个参数非常重要,因为如果参数设置太大或太小,就会导致结果产生偏差并产生负面影响,因此,本发明提出了一种参数设计方案。具体的方法是对第二次聚类的预测结果和第一次聚类得到的伪标签进行评分,即ari:
[0104][0105]
ari是调整兰德系数,ri是兰德系数,expected(ri)是兰德系数的期望值,max(ri)表示兰德系数的最大值,兰德系数ri表达式为:n表示实例总数,用c表示实际的类别划分,k表示聚类结果。定义a为在c中被划分为同一类,在k中被划分为同一簇的实例对数量。定义b为在c中被划分为不同类别,在k中被划分为不同簇的实例对数量。显然ri的值越大说明聚类效果越好;
[0106]
两个簇的结果通过ari评分进行比较,设置γ的取值范围作为比较范围,得分高的结果作为γ参数选择的依据,这样可以从复杂网络的角度对聚类模型进行约束,进一步提高其性能。
[0107]
实施案例2
[0108]
为了验证本发明的有效性,二次聚类后,再次进行了测试。
[0109]
其中,将leiden聚类的分辨率设置到0.1至0.9的范围内,每次更新的步长为0.1,将每次的结果打分后选出最高分数所代表的分辨率参数值,并将其作为进一步聚类的分辨
率参数,然后进行与原来结果的比较,发现聚类的准确性得到了进一步的提升。
[0110]
如图3所示,scvabgme是本发明的方法,further clustering是二次聚类,scale,scvi,cicero是其他另外三种方法,forebrain,insilico,splenocyte,leukemia,gm12878/hl-60,gm12878/hek293t分别是6个sc-atac的单细胞数据集。结果表明,二次聚类后,结果准确率显著提高。
[0111]
图4分别是在forebrain,insilico,splenocyte,leukemia,gm12878/hl-60,gm12878/hek293t等6个单细胞数据集上的聚类结果图,图的右侧分别显示出了识别到的细胞类型。
[0112]
以上所述仅为本发明的优选实施例而已,并不用于限制本发明,对于本领域的技术人员来说,本发明可以有各种更改和变化。凡在本发明的精神和原则之内,所作的任何修改、等同替换、改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。