1.本发明涉及一种高产组氨酸基因工程大肠杆菌菌株及其构建方法,以及使用其菌株发酵生产组氨酸的方法。本发明还涉及用于从发酵过程中回收组氨酸的方法。属于医药、食品和饲料领域。
背景技术:
2.l-组氨酸(l-histidine),工业上一般为盐酸盐形式。盐酸组氨酸为无色结晶或白色结晶性粉末;几乎无臭,味微咸;水溶液显酸性反应。易溶于水,不溶于乙醇、乙醚或氯仿。熔点249~253℃。
3.l-组氨酸传统的制备方法是以血红蛋白为原料,盐酸水解后用离子交换树脂进行柱层析分离,再用盐酸重结晶而得。常规手段是:从猪血、牛血中提取。猪血经喷雾干燥制得血粉,经盐酸水解后,将含有l-组氨酸的洗脱液,浓缩至出现结晶,趁热用盐酸调至ph为2.5,立即加入2倍于溶液量的乙醇,静置,沉淀,过滤,即得l-组氨酸盐酸盐粗品,经脱色、重结晶、干燥得成品。
4.近年来,l-组氨酸的生产工艺主要采用动物毛发或羽毛蛋白为原料。然而,存在动物源性病毒,如牛绵状脑病、禽流感病毒等动物原料带来的其它病毒的风险,因此,人们越来越倾向于寻找使用非动物源原料生产的产品。
5.也有人试图从植物原料(如大豆)的水解产物中提取l-组氨酸,但由于产量低、成本高,脱脂大豆生产l-组氨酸难以实现大量生产,目前仍未能推向商业化。
6.日本味之素株式会社曾公开一项专利,介绍过一项通过微生物发酵法制造l-组氨酸的新方法。该专利的发明人仓桥修等在研究中发现含有从有组氨酸拮抗剂耐性的芽孢杆菌属变异株染色体获得的,装入与组氨酸拮抗剂耐性有关的遗传因子的媒介物的芽孢杆菌属细菌可以高收率生产l-组氨酸。另外,2016年1月,日本食品安全委员会第591次食品安全会对食品添加剂“利用his-no.2菌株生产的l-组氨酸盐酸盐”进行了健康影响评估,总结出了健康影响评估结果(案)。his-no.2菌株是株式会社味之素以escherichia coli k-12来源的突然变异菌株为宿主,导入e.coli k-12菌株来源的l-组氨酸合成相关基因而构建。该公司在获得高产菌种的基础上,利用微生物发酵技术大规模商业化生产l-组氨酸,并研制相应的发酵和纯化技术工艺。
7.本发明是根据微生物代谢工程原理,首先删除微生物内源性嘌呤阻抑蛋白(purr),解除purr对pura表达抑制,提高atp补缺合成速度。其次,筛选对l-组氨酸负反馈抑制不敏感的atp磷酸核糖基转移酶(atp phosphoribosyl transferase,hisg)突变体,并过量表达以提高微生物中atp磷酸核糖基转移酶的作用,达到使微生物以更高效率和更高产量生产l-组氨酸。最后,通过摇瓶、发酵罐发酵优化工艺,并采用陶瓷膜、超滤等膜过滤工艺,去除菌体蛋白、色素、盐类,然后分离提取l-组氨酸盐酸盐。新工艺不仅可以保证工艺和产品的安全性,同时可以提供高质量的l-组氨酸供给市场。
技术实现要素:
8.本发明针对组氨酸的代谢途径,采用新的遗传修饰方法改造微生物,并利用该微生物以更高效率和更高产量生产组氨酸。
9.具体而言,本发明通过筛选atp磷酸核糖基转移酶的基因突变体以解除l-组氨酸对hisg活性的负反馈限制,并过量表达该突变体酶,以增加l-组氨酸合成。同时,删除内源性嘌呤阻抑蛋白(purr),解除purr对pura表达抑制,提高atp补缺合成速度,从而使微生物以更高效率和更高产量生产组氨酸。
10.根据本发明的一个实施方案,本发明涉及一种通过微生物发酵生产组氨酸的方法,该方法包括:
11.a)在发酵培养基中培养微生物,所述微生物包含至少一种能提高微生物中atp磷酸核糖基转移酶作用的遗传修饰;和
12.b)收集从培养步骤a)中产生的组氨酸。
13.在本发明中,提高微生物中atp磷酸核糖基转移酶作用的遗传修饰选自a)微生物中atp磷酸核糖基转移酶的酶活性增加;和/或b)微生物中atp磷酸核糖基转移酶被过量表达;
14.本领域技术人员可以理解,为提高微生物中atp磷酸核糖基转移酶的作用,可以通过筛选编码具有atp磷酸核糖基转移酶的酶活性增加的基因突变体来实现。筛选hisg基因突变体可以通过易错pcr技术得到高频突变基因来完成。为提高微生物中hisg的作用,也可以通过增加其基因拷贝数、更换比天然启动子有更高表达水平的启动子等方式过量表达hisg来实现。
15.在具体的实施方案中,微生物用至少一种包含至少一种能提高微生物中atp磷酸核糖基转移酶作用的遗传修饰的重组核酸分子转化。
16.在一个优选的实施方案中,微生物用至少一种包含编码atp磷酸核糖基转移酶的核酸序列的重组核酸分子转化。
17.在一个方面,编码atp磷酸核糖基转移酶的核酸序列含有至少一种增加atp磷酸核糖基转移酶的酶活性的遗传修饰;进一步优选,所述遗传修饰包括在对应于氨基酸序列seq id no:12的下述位置处的取代中的一种或多种:第232位组氨酸被亮氨酸取代、第252位苏氨酸被丝氨酸取代和第271位谷氨酸被甘氨酸取代;更优选,编码atp磷酸核糖基转移酶的核酸序列为seq id no:11。
18.在另一个方面,所述的atp磷酸核糖基转移酶具有与seq id no:12的氨基酸序列至少约30%相同,优选至少约50%相同,进一步优选至少约70%相同,进一步优选至少约80%相同,更进一步优选至少约90%相同,最优选至少约95%相同的氨基酸序列,其中所述的atp磷酸核糖基转移酶具有酶活性;进一步优选,所述的atp磷酸核糖基转移酶具有seq id no:12的氨基酸序列。
19.在另一个方面,重组核酸分子中编码atp磷酸核糖基转移酶的基因拷贝数增加。
20.在另一个方面,重组核酸分子中包含内源性天然启动子或具有比内源性天然启动子更高表达水平的启动子;优选,具有比内源性天然启动子更高表达水平的启动子选自hce启动子、gap启动子、trc启动子、t7启动子;进一步优选,具有比内源性天然启动子更高表达水平的启动子为trc启动子。
21.在另一个优选的实施方案中,微生物包括至少一种对编码atp磷酸核糖基转移酶的基因的内源性天然启动子的遗传修饰;优选,编码atp磷酸核糖基转移酶的基因的内源性天然启动子被具有更高表达水平的启动子替换;进一步优选,具有更高表达水平的启动子选自hce启动子、gap启动子、trc启动子、t7启动子;最优选,具有更高表达水平的启动子为trc启动子。
22.在本发明中,重组核酸分子转化微生物,选自游离型(即重组核酸分子被装入质粒中)和整合型(即重组核酸分子被整合到微生物的基因组中)。优选,重组核酸分子被整合到微生物的基因组中。
23.根据本发明的优选实施方案,所述微生物进一步包含至少一种能降低微生物中嘌呤阻抑蛋白purr作用的遗传修饰。
24.在上述实施方案的一个方面中,降低微生物中嘌呤阻抑蛋白purr作用的遗传修饰包括但不限于:编码微生物中嘌呤阻抑蛋白purr的内源性基因的部分或完全缺失、或部分或完全失活,和/或编码微生物中嘌呤阻抑蛋白purr基因的内源性天然启动子的部分或完全缺失、或部分或完全失活。优选,降低微生物中嘌呤阻抑蛋白purr作用的遗传修饰为编码微生物中嘌呤阻抑蛋白purr的内源性基因完全缺失,即被删除。在具体的实施方案中,微生物用至少一种包含至少一种能降低微生物中嘌呤阻抑蛋白purr作用的遗传修饰的重组核酸分子转化。
25.在上述任意实施方案的一个方面中,所述重组核酸分子的表达是可诱导的,包括但不限于被乳糖诱导。例如,通过在培养液中添加乳糖等可实现被乳糖诱导表达。
26.本领域技术人员可以理解,本发明中可以使用本领域已知的各种常规发酵培养基。在一个方面中,发酵培养基中包含碳源。在另一个方面中,发酵培养基中包含氮源。在另一个方面中,发酵培养基中包含碳源和氮源。在另一个方面中,发酵培养基中包含碳源、氮源和无机盐。
27.本领域技术人员可以理解,本领域已知的各种碳源均可用于本发明,包括有机碳源和/或无机碳源。优选,碳源选自葡萄糖、果糖、蔗糖、半乳糖、糊精、甘油、淀粉、糖浆和糖蜜中的一种或多种。优选,碳源的浓度维持在约0.1%-约5%。本领域技术人员可以理解,本领域已知的各种氮源均可用于本发明,包括有机氮源和/或无机氮源。优选,氮源选自氨水、氯化铵、硫酸铵、硝酸铵、醋酸铵、硝酸钠、尿素、酵母浸膏、肉类浸膏、蛋白胨、鱼粉、豆粉、麦芽、玉米浆和棉籽粉中的一种或多种。
28.优选,本发明采用补料发酵法。根据本发明的一个方面,补糖液包含葡萄糖,优选,葡萄糖浓度为10%-85%(w/v),进一步优选,葡萄糖浓度为55%-75%(w/v)。
29.在优选的实施方案中,在约20℃-约45℃进行所述培养步骤,进一步优选,在约33℃-约37℃进行所述培养步骤。
30.在优选的实施方案中,在约ph4.5-约ph8.5进行所述培养步骤。进一步优选,在约ph6.7-ph7.2进行所述培养步骤。
31.本领域技术人员可以理解,本发明中可以使用本领域已知的各种常规方法收集组氨酸。优选,可以从发酵培养基中的胞外产物中收集组氨酸。进一步优选,该收集步骤包括选自如下的步骤:(a)从去除微生物的发酵液中沉淀组氨酸;和/或(b)从去除微生物的发酵液中结晶组氨酸。
32.根据本发明,收集步骤进一步包括将发酵液脱色的步骤。脱色步骤可以包括但不限于在对发酵液进行沉淀或结晶之前、在对发酵液进行一次或多次沉淀或结晶重溶解之后进行,脱色包括活性炭处理和/或色谱脱色。所述色谱脱色包括使所述发酵液与离子交换树脂接触的步骤,离子交换树脂包括但不限于阴离子交换树脂和/或阳离子交换树脂,例如使发酵液与阴离子和阳离子交换树脂的混合床接触。
33.在本发明中,微生物可以是任意的微生物(例如细菌、原生生物、藻类、真菌或其它微生物)。在优选的实施方案中,微生物包括但不限于细菌、酵母或真菌。优选,所述的微生物选自细菌或酵母。进一步优选,细菌包括但不限于选自埃希氏菌属(escherichia)、芽孢杆菌属(bacillus)、乳杆菌属(lactobacillus)、假单胞菌属(pseudomonas)或链霉菌属(streptomyces)的属的细菌;更优选,细菌包括但不限于选自大肠杆菌(escherichia coli)、枯草芽孢杆菌(bacillus subtilis)、地衣芽孢杆菌(bacillus licheniformis)、短乳杆菌(lactobacillus brevis)、铜绿假单胞菌(pseudomonas aeruginosa)或浅青紫链霉菌(streptomyces lividans)的种的细菌。进一步优选,酵母包括但不限于选自糖酵母属(saccharomyces)、裂殖糖酵母属(schizosaccharomyces)、念珠菌属(candida)、汉逊酵母属(hansenula)、毕赤酵母属(pichia)、克鲁维酵母属(kluveromyces)和红法夫属(phaffia)的酵母;更优选,酵母包括但不限于选自酿酒酵母(saccharomyce scerevisiae)、粟酒裂殖酵母(schizosaccharo mycespombe)、白色念珠菌(candida albicans)、多形汉逊酵母(hansenulapolymorpha)、巴氏毕赤酵母(pichia pastoris)、加拿大毕赤酵母(pichia canadensis)、马克斯克鲁维酵母(kluyveromyces marxianus)或红法夫酵母(phaffia rohodozyma)。优选,所述的微生物为真菌;进一步优选,真菌包括但不限于选自曲霉属(aspergillus)、犁头霉属(absidia)、根霉属(rhizopus)、金孢子菌属(chrysosporium)、脉孢霉属(neurospora)或木霉属(trichoderma)的属的真菌;更优选,真菌包括但不限于选自黑曲霉(aspergillus niger)、构巢曲霉(aspergillus nidulans)、蓝色犁头霉(absidia coerulea)、米根霉(rhizopus oryzae)、劳肯诺温斯金孢子菌(chrysosporium lucknowense)、粗糙脉孢霉(neurospora crassa)、间型脉孢霉(neurospora intermedia)或里氏木霉(trichoderma reesei)。特别优选的大肠杆菌菌株包括k-12、b和w,最优选k-12。尽管大肠杆菌作为优选的微生物且用作本发明各种实施方案的实例,但是应理解在本发明方法中可以使用产生组氨酸且可以通过遗传修饰以提高组氨酸产量的任意其它微生物。用于本发明的微生物也可以称作生产生物体。
34.在本发明中,术语组氨酸均指l-组氨酸。术语l-组氨酸可以称作(s)-2-氨基-3-(4-咪唑基)丙酸,l-α-氨基-β-4-咪唑基丙酸。术语l-组氨酸可以分别缩写为l-his。
35.术语提高微生物中酶的作用是指微生物中该酶的活性增加和/或该酶被过量表达,从而提高了微生物中由该酶所催化的底物生成产物的量。
36.术语降低微生物中酶的作用是指微生物中该酶的活性降低和/或该酶的表达减少,从而降低了微生物中由该酶所催化的底物生成产物的量。
37.术语酶活性增加是指酶催化一定化学反应的能力增加。其涵盖了在酶受产物抑制作用和酶对底物亲合力不变的情况下酶自身催化化学反应的能力增加,和/或由于酶受产物抑制作用降低导致和/或酶对底物亲合力增加所导致的酶催化化学反应的能力增加。术语酶受产物抑制作用降低是指催化反应的酶的活性受其终产物特异抑制作用而降低。术语
酶对底物亲合力增加是指酶对所催化的底物的亲合力增加。
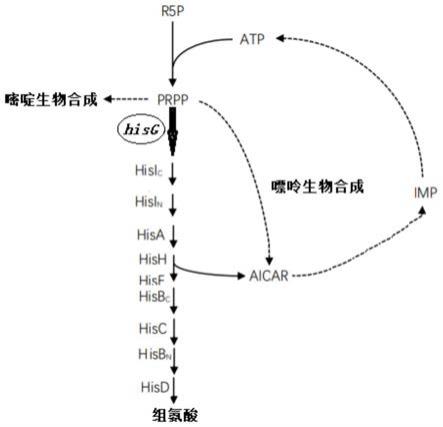
38.图1以大肠杆菌为例解释了本发明公开的用于大规模生产组氨酸代谢途径中遗传修饰的主要方面。图1公开了用于合成组氨酸的方法,包括对hisg的修饰。本领域技术人员可以理解,其它微生物具有类似的糖代谢途经,且在这类途经中基因和蛋白质具有类似的结构和功能。因此,本发明所讨论的除适用于大肠杆菌外同样适用于其它微生物,且其它微生物显然包括在本发明中。
39.本领域中已知具有相同生物活性的酶可以具有不同的名称,这取决于该酶来源于什么样的微生物。下面是本文涉及的许多酶的可选名称和来自一些生物体的编码这类酶的具体基因名称。这些酶的名称可以互换使用或如果合适用于给定的序列或生物体,但本发明意图包括来自任意生物体的指定功能的酶。
40.例如,本文一般称作“atp磷酸核糖基转移酶”的酶,即指atp依赖性的磷酸核糖基转移酶,是l-组氨酸合成途径上从的第二个酶,是整个反应一个关键酶,同时也受终产物l-组氨酸的反馈抑制。解除l-组氨酸对atp磷酸核糖基转移酶活性的负反馈限制,可增加l-组氨酸合成。来自大肠杆菌(escherichia coli)的atp磷酸核糖基转移酶(atp-phosphoribosyl transferase)一般称作hisg。来自各种生物体的atp磷酸核糖基转移酶是本领域中公知的,且可用于本发明遗传改造策略中。例如,本文描述了来自大肠杆菌的atp磷酸核糖基转移酶具有由seq id no:7表示的氨基酸序列。
41.例如,本文一般称作“嘌呤阻抑蛋白”的酶,可通过与pur操纵子调控区dna的相互作用来抑制该操纵子的转录。删除内源性嘌呤阻抑蛋白,解除purr对pura表达抑制,提高atp补缺合成速度。来自大肠杆菌(escherichia coli)的嘌呤阻抑蛋白一般称作purr。例如,本文描述了来自大肠杆菌的嘌呤阻抑蛋白具有由seq id no:4表示的核苷酸序列。
[0042]“trc启动子”经过巧妙的设计可用于原核表达,例如大肠杆菌表达系统。trc启动子是本领域中公知的且可用于本发明的遗传改造策略中。例如,本文描述了的trc promoter具有seq id no:13表示的核苷酸序列。
[0043]
本发明的有益效果在于:本发明证实可通过筛选atp磷酸核糖基转移酶的基因突变体以解除l-组氨酸对hisg活性的负反馈限制,并过量表达以提高微生物中atp磷酸核糖基转移酶的作用,增加l-组氨酸合成。同时删除内源性嘌呤阻抑蛋白,解除purr对pura表达抑制,提高atp补缺合成速度,从而使该微生物以更高效率和更高产量生产组氨酸。
[0044]
将本文引述或描述的各公开文献和参考文献的全部内容引入本文作为参考。
附图说明
[0045]
图1为大肠杆菌中组氨酸生物合成与代谢途径中遗传修饰。
具体实施方式
[0046]
下文将结合具体实施例对本发明做更进一步的详细说明。下列实施例仅为示例性地说明和解释本发明,而不应被解释为对本发明保护范围的限制。凡基于本发明内容所实现的技术均涵盖在本发明旨在保护的范围内。
[0047]
除非另有说明,实施例中使用的原料和试剂均为市售商品。
[0048]
实施例1
[0049]
本实施例描述了通过删除内源性嘌呤阻抑蛋白(purr),以降低微生物中嘌呤阻抑蛋白作用的遗传修饰。
[0050]
嘌呤阻抑蛋白purr通过与pur操纵子调控区dna的相互作用来抑制该操纵子的转录。删除内源性嘌呤阻抑蛋白purr基因序列以解除purr对pura表达抑制,提高atp补缺合成速度,从而提高pur操纵子表达调控。所述亲本菌株zas-001是已经部分遗传操作处理的大肠杆菌菌株。
[0051]
1、制备red重组打靶用线性dna全长pcr片段
[0052]
(1)pcr扩增fkanrf片段
[0053]
fkanrf片段,即frt-kanr-frt片段,是指将卡拉霉素抗性基因(kanr)的两端装有flp重组酶特异性识别的frt位点碱基序列。
[0054]
设计引物:正向引物(mfkanf-f)seq id no:1,反向引物(mfkanf-r)seq id no:2。
[0055]
模板:ppic9k。
[0056]
pcr反应条件:第一步:94℃变性1min;第二步:94℃30s,55℃30s,72℃40s,循环30次;第三步:72℃延伸10min。
[0057]
fkanrf大小:1.28kb。其核苷酸序列seq id no:3。
[0058]
pcr产物经1%琼脂糖凝胶电泳分离、纯化回收片段。
[0059]
(2)pcr扩增red重组打靶用线性dna全长片段
[0060]
设计同源臂引物:根据purr核苷酸序列seq id no:4,设计删除purr序列的同源臂正向引物(purrko-f)seq id no:5,反向引物(purrko-r)seq id no:6。
[0061]
模板:扩增的fkanrf pcr片段。
[0062]
pcr反应条件:第一步:94℃变性1min;第二步:94℃30s,55℃30s,72℃40s,循环30次;第三步:72℃延伸10min。
[0063]
扩增产物:同源臂 fkanrf 同源臂。
[0064]
将pcr产物琼脂糖凝胶电泳分离、纯化回收,得到100ng/μl的线性dna全长pcr片段用于red重组打靶。
[0065]
2、red重组操作
[0066]
首先,将pkd46载体转入亲本菌株大肠杆菌zas-001菌株中。然后,电转化制备好的打靶用线性dna片段,筛选阳性克隆。最后,消除抗性基因。
[0067]
(1)转化pkd46质粒
[0068]
pkd46载体是带有表达red重组酶基因的质粒,表达exo、bet和gam三基因片段,3个基因置于阿拉伯糖启动子下,经l-阿拉伯糖诱导就可以大量表达。为达到通过red重组修饰染色体上目标基因的目的,有必要将pkd46质粒转化到大肠杆菌中。
[0069]
1)感受态制备:首先,将保存于-20℃的大肠杆菌zas-001菌液,按1:50-100接种于10ml lb液体培养基中,37℃,225rpm,振荡培养2-3小时。再将培养液加入到10ml离心管中,4000g
×
5min,弃去上清,用冰浴的0.1m cacl
2 5ml悬浮5min。最后,4000g
×
5min离心,弃去上清,用冰浴的0.1m cacl25ml悬浮。-4℃静置12小时,自然沉降。其中,0.1m cacl2的制备:用无水cacl2配1m的cacl2,用蒸气压力为15lbf/in2的高压灭菌20min,分装1.5ml于-20℃保存,用时融化后按1:10比例稀释配成0.1m的cacl2。
[0070]
2)质粒转化:取自然沉降的菌体250μl,加入5μl pkd46质粒,-4℃,30min。然后,42
℃水浴1.5min,加入soc培养基0.7ml,30℃摇2小时。取0.2ml菌液,涂氨苄青霉素(amp)平板。30℃过夜(12-16小时)培养。挑单克隆,加入5ml lb液体培养基中培养,抽质粒鉴定。保存阳性菌种备用。
[0071]
(2)电转化制备好的打靶用线性dna片段,筛选阳性克隆
[0072]
1)电转感受态的制备:将含pkd46大肠杆菌zas-001菌种接种于含有氨苄青霉素lb培养基的试管,250rpm摇床过夜,第二天以1%的量接种至含有amp的lb培养基中,30℃培养,待od
600
达到0.2左右后,加入0.2%的l-阿拉伯糖,30℃诱导35分钟,直至od
600
达到0.4左右。冰浴冷却。用超纯水洗一次,10%甘油洗两次,最后用10%甘油重悬,甘油用量以使菌体被浓缩500-1000倍的终浓度为宜。
[0073]
2)电击转化:将2mm电转杯从70%乙醇中取出,用灭菌超纯水洗2次,紫外灯照射30分钟。4℃预冷30分钟。取90μl最终重悬的细胞,移至预冷的离心管,加入5μl(100ng以上)步骤(1)得到的全长pcr片段(线性dna),用枪轻轻吸打混匀,冰浴30分钟。电转参数:2500v,200ω,25μf。
[0074]
3)复苏与筛选阳性克隆:加入1ml的lb液体培养基,37℃,100rpm,1小时。然后每200μl涂布一个卡拉霉素(kan)平板,共5个。均匀、涂干。30℃培养24个小时。挑在卡拉霉素抗性下生长的克隆,作pcr鉴定,筛选阳性克隆。
[0075]
所获得菌种编号:zas-002(zas-001,
△
purr::fkanrf)。
[0076]
3、抗性基因的消除
[0077]
为便于后续工作,可消除所获得菌种(阳性克隆)中的抗性基因。消除抗性基因可借助pcp20质粒完成。pcp20是带有氨苄青霉素和氯霉素抗性基因的质粒,热诱导后可表达flp重组酶,该酶可特异性识别frt位点,通过重组可将frt位点间的序列删除,只保留一个frt位点。
[0078]
将pcp20转入上述卡拉霉素抗性克隆,30℃培养8h,后提高到42℃过夜,热诱导flp重组酶表达,质粒逐渐丢失。用接种环蘸菌液在无抗生素培养基上划板,挑长出的单克隆点到卡拉霉素抗性平板上,未生长的为卡拉霉素抗性基因已被flp重组酶删除的克隆。用鉴定引物作pcr对卡拉霉素抗性消失的克隆进行鉴定。
[0079]
所获得菌种编号:zas-003(zas-001,
△
purr)。
[0080]
实施例2
[0081]
本实施例描述了atp磷酸核糖基转移酶基因hisg克隆、基因突变体筛选、亲本菌株转化。
[0082]
atp磷酸核糖基转移酶hisg是l-组氨酸合成途径上的第一个酶,是整个反应一个关键酶,同时也受终产物l-组氨酸的反馈抑制。通过扩增大肠杆菌hisg基因,置于trc启动子控制下转化菌种,使之过量表达,期望增加是l-组氨酸合成。为了进一步提高生产菌株中的组氨酸合成量,筛选编码具有酶活性增加的atp磷酸核糖基转移酶的基因突变体。解除l-组氨酸对hisg活性的负反馈限制。为了达到该目的,用易错pcr技术扩增克隆的基因,通过用于扩增的dna聚合酶,在导致高频错配的条件下扩增所述基因,以便在pcr产物中得到高频突变。
[0083]
1、大肠杆菌hisg基因的克隆与重组菌制备
[0084]
根据ncbi查找nc_007779.1,获得大肠杆菌hisg基因氨基酸序列seq id no:7。按
大肠杆菌偏爱碱基密码子进行碱基优化,其核苷酸序列seq id no:8。将seq id no:8基因合成,并在5’端装入nco i酶切点序列,在3’端装入hindiii酶切点序列,与puc57-t载体连接并测序鉴定,获得质粒hisg/puc57。
[0085]
1)重组质粒制备:用ncoi和hindiii分别酶切质粒hisg/puc57和载体ptrc99a,琼脂糖凝胶电泳分离、纯化回收hisg片段和ptrc99a片段,用t4dna连接酶,16℃将连接过夜,并鉴定,得到重组质粒hisg/ptrc99a。
[0086]
2)感受态制备:首先,将保存于-20℃的zas-003冻存菌液,按1:50-100接种于10ml lb液体培养基中,37℃,225rpm,振荡培养2-3小时。再将培养液加入到10ml离心管中,4000g
×
5min,弃去上清,用冰浴的0.1m cacl
2 5ml悬浮5min。最后,4000g
×
5min离心,弃去上清,用冰浴的0.1m cacl
2 5ml悬浮。-4℃静置12小时,自然沉降。
[0087]
3)质粒转化:取自然沉降的菌体250μl,加入5μl hisg/ptrc99a质粒,-4℃,30min。然后,42℃水浴1.5min,加入soc培养基0.7ml,30℃摇2小时。取0.2ml菌液,涂青霉素平板。30℃过夜(12-16小时)培养。挑单克隆,加入5ml lb液体培养基中培养,抽质粒鉴定。保存阳性菌种备用。得到将hisg/ptrc99a转化至zas-003的重组菌,命名为:zas-004(zas-001,hisg/ptrc99,
△
purr)。
[0088]
2、易错pcr扩增短双歧杆菌atp磷酸核糖基转移酶基因hisg
[0089]
利用taq dna聚合酶不具有3'-5'校对功能的性质,在高镁离子浓度(8mmol/l)和不同浓度dntp的浓度下(其中,datp和dgtp浓度为1.5mmol/l;dttp和dctp浓度为3.0mmol/l),来控制随机突变的频率,向目的基因中引入随机突变,构建突变库;模板浓度a260值为1000ng/ml,酶浓度为5u/μl,引物浓度为100μm。
[0090]
易错pcr反应体系(50μl):10
×
pcr反应缓冲液5μl,dntp(2.5mm)5μl,mgcl2(2.5mm)5μl,正向引物(hisg-f,seq id no:9)1μl,反向引物(hisg-r,seq id no:10)1μl,dna模板(hisg/puc57)0.1μl,taq dna聚合酶0.5μl,ddh2o 32.4μl。
[0091]
pcr程序:96℃预变性4min;94℃变性1min,56℃退火1min,75℃延伸2min,45个循环;最后75℃延伸15min,采用胶回收方法回收pcr产物(产物大小:0.9kb);取5μl产物1%琼脂糖凝胶电泳检验,-20℃保存备用。
[0092]
3、构建atp磷酸核糖基转移酶的基因突变体库
[0093]
将上述pcr产物经限制性内切酶nco i和hind iii双酶切消化后,与用nco i和hind iii内切酶消化的ptrc99a质粒进行连接反应,然后用连接产物混合物转化大肠杆菌top10,获得大量克隆转化子,构建转化菌体突变库。
[0094]
4、筛选高酶活突变体及其重组菌制备
[0095]
从转化菌体突变库中,随机挑取突变克隆约5000株,以野生型zas-004为对照,分别接种至含50μg/ml青霉素(amp)的5ml lb培养基中,37℃、150rpm培养18h后,10000rpm,5mim离心收集菌体。弃上清后,在4℃下重悬于1ml pbs(ph值7.5,10mmol/l)溶液中,在冰浴条件下选取300v电压,超声3s间歇6s对其进行超声破碎10min,离心取上清作为酶粗提液,进行酶活测定。
[0096]
atp磷酸核糖基转移酶的活性检测:以底物5-磷酸核糖-α-焦磷酸(5-phosphorlbosylα-pyrophosphate,prpp)减少为测定标记。酶活单位定义:在酶促反应条件下,每分钟减少相当于1μmol prpp所需的酶量,定义为一个酶活力单位(iu)。具体操作如
下:以5ml反应体系为酶活测定体系,其中含500mmol/l prpp、5mmol/l葡萄糖、100mmol/l tris-hcl(ph8.0)及100μl粗酶液。酶活反应在37℃水浴中进行,保温4h,然后将酶解液在70℃下10min终止反应。3000rpm离心10min,取上清液。hplc测定prpp含量。
[0097]
结果表明:最高突变体菌株的酶活为77.3iu/ml,对照菌株的酶活为15.4iu/ml。通过易错pcr对hisg进行改造,获得酶活力提高约5倍的突变株。挑选该酶活性最高的突变体菌株,提取质粒测序。结果表明:该atp磷酸核糖基转移酶突变体基因序列如seq id no:11所示,对应的氨基酸序列如seq id no:12所示。与野生型的atp磷酸核糖基转移酶基因序列比对,共发生了5处碱基点突变:396g/a,591g/a,695a/t,754a/t,822a/g;致氨基酸3处错义突变,其突变点分别为:h232l(第232位组氨酸变为亮氨酸),t252s(第252位苏氨酸变为丝氨酸),e271g(第271位谷氨酸变为甘氨酸)。将该突变基因命名为hisgm。
[0098]
按上述同样方法,制备hisgm/ptrc99a质粒。然后,将hisgm/ptrc99a质粒转化入菌株zas-003,获得重组菌命名为:zas-005(zas-001,hisgm/ptrc99,
△
purr)。
[0099]
实施例3
[0100]
本实施例描述重组菌摇瓶发酵试验、产物测定,及其组氨酸产量
[0101]
1、发酵培养液制备
[0102]5×
m9培养基配制:将在约800ml双蒸水(ddh2o)中加入64g na2hpo4·
7h2o、15g kh2po4、2.5g nacl、5.0g nh4cl,溶解后,加水至1000ml。121℃灭菌30分钟。再分别配制1m mgso4、1m cacl2、20%葡萄糖,并单独灭菌。然后按表1配制m9培养液,其中,1000
×
微量元素溶液按表2配制。
[0103]
表1.m9培养液成分
[0104]
成分用量(ml/l)5
×
m92001m mgso421m cacl20.120%葡萄糖201000
×
微量元素溶液1ddh2o至1000ph6.9
[0105]
表2.1000
×
微量元素溶液成分
[0106]
成分用量(g/l)cocl2·
6h2o0.01cuso4·
5h2o0.01mnso4·
h2o0.033fe so4·
7h2o0.50znso4·
7h2o0.38h3bo30.01namoo4·
2h2o0.01ph3
[0107]
2、重组菌zas-005与对照zas-004、zas-003、zas-001摇瓶发酵试验
[0108]
分别取lb平板培养基上新鲜培养的重组菌zas-005与对照zas-004、zas-003、亲本菌株zas-001的单克隆菌株,接种于3ml的lb液体培养基试管(13
×
150mm)中,30℃,225rpm,培养约8小时。lb液体培养基成分:5g/l酵母粉,10g/l蛋白胨,10g/l nacl。然后取种子培养液,3%接种于含50ml的发酵培养液(m9培养液)250ml摇瓶中。起始od
600
约0.5,37℃下,225rpm培养,发酵周期72小时。在第24小时、48小时,用10m naoh调节发酵液ph至7.0。根据发酵液糖耗情况,分次加入65%葡萄糖液维持葡萄糖浓度在20g/l。发酵结束,取1ml发酵液,离心。用hplc法测定组氨酸含量。
[0109]
3、组氨酸含量的hplc法测定
[0110]
取样品,稀释10倍,摇匀,0.22um膜过滤。
[0111]
流动相:乙腈:水(含0.5%磷酸二氢钠)=10:90
[0112]
色谱柱:c18
[0113]
波长:210nm
[0114]
流速:1ml/min
[0115]
进样量:20μl
[0116]
出峰时间:5.2min
[0117]
4、重组菌质粒转化对摇瓶发酵组氨酸产量的影响
[0118]
摇瓶发酵产量情况见表3。结果表明:对照菌种zas-001、zas-003未检出,重组菌zas-004产量很低,重组菌zas-005产量明显提高。
[0119]
表3.ptrc-hisgm基因盒整合重组菌摇瓶发酵产量
[0120]
菌种组氨酸产量(g/l)zas-0051.9
±
0.3zas-0040.2
±
0.1zas-0030.0
±
0.0zas-0010.0
±
0.0
[0121]
以上结果显示:在删除内源性嘌呤阻抑蛋白后,atp磷酸核糖基转移酶超表达可提高l-组氨酸产量;通过易错pcr技术筛选突变体也可大大提高l-组氨酸产量,这可能是由于所获得的该酶突变体解除l-组氨酸负反馈限制,酶活性增加所致。
[0122]
实施例4
[0123]
本实施例描述组氨酸分离纯化后处理工艺。
[0124]
以重组工程菌株zas-005作为生产菌种,在12m3发酵罐按常规工艺发酵。发酵结束后,产物分离纯化工艺具体如下:
[0125]
发酵液用陶瓷膜过滤,固液分离。母液加热至70℃,用6mol/l hcl调ph至7.5,加活性炭脱色,保温搅拌1h。脱色后透光率要达到99%以上。板框过滤,滤液经超滤后,85℃真空浓缩,至有大量结晶出现,冷析,离心收集组氨酸,即得组氨酸精品,母液回收,再利用。
[0126]
组氨酸精品置于双锥旋转蒸发干燥器内干燥,至水分达标。产物:l-组氨酸盐酸盐成品。
[0127]
虽然,上文中已经用一般性说明及具体实施方案对本发明作了详尽的描述,但在本发明基础上,可以对之作一些修改或改进,这对本领域技术人员而言是显而易见的。因此,在不偏离本发明精神的基础上所做的这些修改或改进,均属于本发明要求保护的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。