1.本发明属于有机合成领域,涉及一种2-氯-3-氰基吡啶的制备方法。
背景技术:
2.吡啶环是含氮环最主要的杂环之一。吡啶环在许多性质和特点上特别在疏水性上和苯环有很大的区别,因此用吡啶环替代苯环得到的新农药化合物往往具有更高的生物活性、内吸性、选择性和更低的毒性,这也是吡啶杂环类农药在杀虫剂、除草剂、杀菌剂等各种农药的各个类别中的研究、开发和应用有很大的发展。
3.最早的含吡啶农药为17世纪末到18世纪初欧洲使用的植物杀虫剂-烟草浸出液,在1690年欧洲已用其防治梨花网蚝。八十年代以来含吡啶农药进入稳定均衡发展时期,每年发表专利几十件,从结构上看,有机磷、氨基甲酸酯、磺酰脲等各类农药都有含吡啶的化合物。
4.卤代吡啶类化合物主要包括卤代元素氟、氯、溴、碘的化合物,其中氯代吡啶作为化学工业(特别是精细化工)的重要原料,主要应用于医药中间体、农药中间体、香料类、染料、表面活性剂、橡胶助剂等多个领域。随着吡啶类化合物市场的不断开拓,研究和生产吡啶氯代化合物已成为非常迫切的需求。
5.2-氯烟酸是重要的医药和农药中间体,通过2-氯烟酸可以合成许多医用抗菌素、治疗心血管疾病的药物以及农用杀菌剂、杀虫剂、除草剂等,其结构式如下:
[0006][0007]
2-氯-3-氰基吡啶是合成2-氯烟酸的重要中间体,通过将2-氯-3-氰基吡啶在碱性条件下进行简单的水解就能够以高收率得到2-氯烟酸,2-氯烟酸结构式如下:
[0008][0009]
目前制备2-氯-3-氰基吡啶通常是以3-氰基吡啶为起始原料,通过氧化反应制得3-氰基吡啶n-氧化物,对3-氰基吡啶n-氧化物进行氯代得到2-氯-3-氰基吡啶,但现有的氯代工艺存在产率低、选择性差的问题,仍无法以高收率和高选择性制备得到2-氯-3-氰基吡啶。
技术实现要素:
[0010]
针对现有技术中存在的的缺陷,本发明提供2-氯-3-氰基吡啶的制备方法,该方法通过对氯化反应体系ph的控制,能够以高选择性和高收率制备得到2-氯-3-氰基吡啶,具有操作简单,条件温和的优点。
[0011]
本发明提供一种2-氯-3-氰基吡啶的制备方法,包括:将3-氰基吡啶n-氧化物溶于三氯氧磷中,在0-5℃下滴加有机碱并控制体系的ph为9.5-10.5,滴加完毕后对体系进行升温处理,得到所述2-氯-3-氰基吡啶。
[0012]
如上所述的制备方法,其中,所述有机碱选自环己胺、三乙胺、二异丙基胺、吡啶、苯胺中的至少一种。
[0013]
如上所述的制备方法,其中,所述3-氰基吡啶n-氧化物与所述有机碱的物质的量之比为1:(3-5)。
[0014]
如上所述的制备方法,其中,所述有机碱的滴加速度为200-300g/h。
[0015]
如上所述的制备方法,其中,所述3-氰基吡啶n-氧化物与所述三氯氧磷的物质的量之比为1:(3.13-4.35)。
[0016]
如上所述的制备方法,其中,所述升温处理包括对所述体系升温至80-90℃进行氯化反应。
[0017]
如上所述的制备方法,其中,所述氯化反应的时间为6-8h。
[0018]
如上所述的制备方法,还包括对所述氯化反应后的反应体系进行分离提纯,所述分离提纯包括以下步骤:
[0019]
1)减压蒸馏除去所述反应体系中的三氯氧磷,得到浓缩液;
[0020]
2)冰水浴下,在所述浓缩液加入去离子水中得到稀释液,搅拌状态下加入碱调节所述稀释液的ph至2-2.5;
[0021]
3)在冰水浴下搅拌3-4h后,抽滤干燥得到所述2-氯-3-氰基吡啶。
[0022]
如上所述的制备方法,其中,所述3-氰基吡啶n-氧化物通过3-氰基吡啶与双氧水反应得到。
[0023]
本发明还提供一种如上所述的制备方法在2-氯烟酸合成中的应用。
[0024]
相比于现有技术,本发明具有以下有益效果:
[0025]
本发明所提供的2-氯-3-氰基吡啶的制备方法以3-氰基吡啶n-氧化物为起始原料,以三氯氧磷为氯化剂对3-氰基吡啶n-氧化物进行氯化反应,通过控制反应体系的ph为9.5-10.5,能够高选择性和高收率制备得到2-氯3-氰基吡啶,该方法具有操作简单,条件温和的优点。
附图说明
[0026]
图1为本发明实施例1制备得到的2-氯-3-氰基吡啶的1h nmr图。
具体实施方式
[0027]
为使本发明的目的、技术方案和优点更加清楚,下面将结合本发明的实施例,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0028]
本发明提供一种2-氯-3-氰基吡啶的制备方法,包括以下步骤:
[0029]
将3-氰基吡啶n-氧化物溶于三氯氧磷中,在0-5℃下滴加有机碱并控制体系的ph为9.5-10.5,滴加完毕后对体系进行升温处理,得到2-氯-3-氰基吡啶。
[0030]
上述制备方法可以用如下反应式表示:
[0031][0032]
本发明以3-氰基吡啶n-氧化物为原料,以三氯氧磷为氯化剂,无需使用溶剂,直接将3-氰基吡啶n-氧化物溶于过量的三氯氧磷中进行氯化反应,并在反应体系中加入有机碱中和氯化反应中生成的酸,在低温下滴加有机碱控制反应的速度,避免过多副产物的生成。
[0033]
发明人研究发现,通过在0-5℃的低温下滴加有机碱并控制反应体系的ph为9.5-10.5,能够使氯化反应高化学选择性地生成在吡啶环的c2位,避免副产物6-氯-3-氰基吡啶的生成。正是基于上述研究发现,从而完成了本发明。
[0034]
进一步的,本发明所使用的有机碱可选自环己胺、三乙胺、二异丙基胺、吡啶、苯胺中的至少一种。
[0035]
上述有机碱都是适用于氯化反应的缚酸剂,其碱性足够与质子酸生成稳定的盐,在反应中能稳定存在,不易发生副反应,反应完成后易于除去,不会增加后处理的难度。
[0036]
在一种具体的实施方式中,3-氰基吡啶n-氧化物与有机碱的物质的量之比为1:(3-5),在此物质的量之比范围内,能够控制反应体系的ph为9.5-10.5,有利于高选择性地生成2-氯-3-氰基吡啶。
[0037]
进一步的,控制有机碱的滴加速度为每小时滴加200-300g,酸碱中和反应会放出大量的热量,因此控制滴加速度能够降低反应速度,避免过多副产物的生成。
[0038]
为使反应原料3-氰基吡啶n-氧化物能够转化地更加完全,可控制3-氰基吡啶n-氧化物与三氯氧磷的物质的量之比为1:(3.13-4.35)。
[0039]
过量的三氯氧磷一方面可作为氯化剂与3-氰基吡啶n-氧化物发生氯化反应,另一方面可作为反应溶剂促进3-氰基吡啶n-氧化物的溶解,从而使3-氰基吡啶n-氧化物在均相状态下更利于反应的进行。
[0040]
待有机碱滴加完毕后,为加快氯化反应的速度,将反应体系升温至80-90℃下反应。
[0041]
反应进行6-8h时,检测反应体系中的原料3-氰基吡啶n-氧化物已经完全反应,可判定反应已完成。
[0042]
反应完成后,为得到高纯度的2-氯-3-氰基吡啶氮氧化物,还需要对氯化反应液进行分离提纯。
[0043]
具体的,本发明中对氯化反应液的分离提纯包括以下步骤:
[0044]
1)减压蒸馏除去反应体系中的三氯氧磷,得到浓缩液;
[0045]
2)冰水浴下,在浓缩液中加入去离子水得到稀释液,搅拌状态下加入碱调节稀释液的ph为2-2.5。
[0046]
3)在冰水浴下搅拌3-4h,抽滤干燥得到2-氯-3-氰基吡啶。
[0047]
步骤1)中,反应后体系中剩余的三氯氧磷可通过减压蒸馏回收,有利于节约反应成本。
[0048]
步骤2)中,通过将浓缩物稀释并调节ph至2-2.5,在此过程中逐渐有固体产品析出。
[0049]
步骤3)中,在冰水浴下继续搅拌3-4h,能够使固体充分析出,抽滤干燥后即可得到2-氯-3-氰基吡啶。
[0050]
分离提纯后,计算出氯化反应的收率可达95%以上,其中,制得的2-氯-3-氰基吡啶的纯度可达99.5%以上。
[0051]
上述制备方法所使用的3-氰基吡啶n-氧化物原料可通过3-氰基吡啶与双氧水反应得到,可通过如下反应式表示:
[0052][0053]
其中,具体操作包括:将3-氰基吡啶加入酸性溶液中回流,达到回流温度后向反应液中缓慢滴加双氧水,滴加完毕后,继续回流2h后检测到3-氰基吡啶已完全转化,将反应液降温放置,待固体完全析出后过滤得到3-氰基吡啶。
[0054]
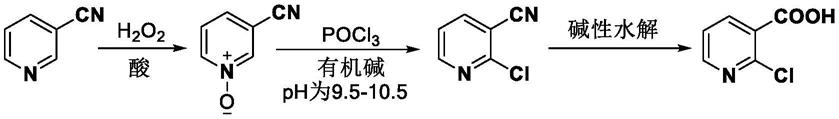
本发明还提供一种2-氯-3-氰基吡啶的制备方法在2-氯烟酸合成中的应用,将此制备方法用于2-氯烟酸的合成中可用如下合成路线表示:
[0055][0056]
通过本发明的制备方法得到2-氯-3-氰基吡啶后,可进一步通过碱性条件水解,得到2-氯烟酸,由于本发明所提供的的制备方法可高选择性和高收率地获得2-氯-3-氰基吡啶,因此可提高2-氯烟酸的生产效率,更适用于工业化生产。
[0057]
以下将结合具体的实施例对本发明所提供的2-氯-3-氰基吡啶的制备方法进行详细地介绍。
[0058]
在下述实施例中,如无特殊说明,使用的方法均是本领域常用的方法,使用的原料均可通过商购或常规方法制备得到。
[0059]
实施例1
[0060]
本实施例2-氯-3-氰基吡啶的制备方法如下:
[0061]
1)3-氰基吡啶n-氧化物的制备
[0062]
在3000ml的四口烧瓶中加入500g(4.80mol)3-氰基吡啶,1600g浓硫酸,升温至90℃至回流,回流状态下向反应体系中缓慢滴加400ml质量浓度为30%的双氧水,滴加完毕后继续回流反应2h,检测原料3-氰基吡啶已完全转化;
[0063]
将反应液降至室温下过滤得到固体产品,50℃下烘干称重,得到548g的3-氰基吡啶n-氧化物,收率为95%。
[0064]
2)2-氯-3-氰基吡啶的制备
[0065]
在3000ml的四口烧瓶中加入500g(4.16mol)3-氰基吡啶n-氧化物和2000g(13.04mol)三氯氧磷,搅拌,在冰水浴0-5℃下,滴加速度为每小时200g滴加3摩尔当量的环己胺,控制反应温度在0-5℃,控制反应体系的ph为9.5-10.5,滴加完毕后,升温至90℃下反应6-8h,检测到3-氰基吡啶n-氧化物小于1%;
[0066]
将反应液降至室温,在60℃下减压蒸馏,顶温45℃下回收得到1300g三氯氧磷。向
剩余的浓缩液中加入1000g冰水稀释,搅拌状态下加入氢氧化钠溶液,调节ph至2-2.5,有大量褐色固体析出,继续在冰水浴下搅拌1h,抽滤干燥得到550g褐色固体,2-氯-3-氰基吡啶的摩尔收率为95%,纯度为99.5%,通过高效液相色谱分析剩余的0.5%杂质为6-氯-3-氰基吡啶。
[0067]
需要说明的是,本实施例中2-氯-3-氰基吡啶的摩尔收率通过抽滤干燥后得到的褐色固体的质量除以理论计算得到的2-氯-3-氰基吡啶的质量得到,在以下实施例和对比例中的2-氯-3-氰基吡啶的摩尔收率的计算方式与本实施例均一致。
[0068]
图1为本发明实施例1制备得到的2-氯-3-氰基吡啶的1h nmr图,如图1所示,实施例1制备得到的2-氯-3-氰基吡啶通过1h nmr表征如下:
[0069]1h nmr(400mhz,cdcl3)δ7.43(dd,1h),8.05(dd,1h),8.63(dd,1h).
[0070]
实施例2
[0071]
本实施例2-氯-3-氰基吡啶的制备方法如下:
[0072]
1)3-氰基吡啶n-氧化物的制备
[0073]
在3000ml的四口烧瓶中加入500g(4.80mol)3-氰基吡啶,1600g浓硫酸,升温至90℃至回流,回流状态下向反应体系中缓慢滴加400ml质量浓度为30%的双氧水,滴加完毕后继续回流反应2h,检测原料3-氰基吡啶已完全转化;
[0074]
将反应液降至室温下过滤得到固体产品,50℃下烘干称重,得到548g的3-氰基吡啶n-氧化物,收率为95%。
[0075]
2)2-氯-3-氰基吡啶的制备
[0076]
在3000ml的四口烧瓶中加入500g(4.16mol),3-氰基吡啶n-氧化物和2000g(13.04mol)三氯氧磷,搅拌,在冰水浴0-5℃下,滴加速度为每小时200g滴加3摩尔当量的吡啶,控制反应温度在0-5℃,控制反应体系的ph为9.5-10.5,滴加完毕后,升温至90℃下反应6-8h,检测到3-氰基吡啶n-氧化物小于1%;
[0077]
将反应液降至室温,在60℃下减压蒸馏,顶温45℃下回收得到1300g三氯氧磷。向剩余的浓缩液中加入1000g冰水稀释,搅拌状态下加入氢氧化钠溶液,调节ph至2-2.5,有大量褐色固体析出,继续在冰水浴下搅拌1h,抽滤干燥得到492g褐色固体,经1h nmr确认得到的固体产物为2-氯-3-氰基吡啶,2-氯-3-氰基吡啶摩尔收率为85%,纯度为95%,通过高效液相色谱分析可知,剩余的5%杂质为6-氯-3-氰基吡啶。
[0078]
实施例3
[0079]
本实施例2-氯-3-氰基吡啶的制备方法如下:
[0080]
1)3-氰基吡啶n-氧化物的制备
[0081]
在3000ml的四口烧瓶中加入500g(4.80mol)3-氰基吡啶,1600g浓硫酸,升温至90℃至回流,回流状态下向反应体系中缓慢滴加400ml质量浓度为30%的双氧水,滴加完毕后继续回流反应2h,检测原料3-氰基吡啶已完全转化;
[0082]
将反应液降至室温下过滤得到固体产品,50℃下烘干称重,得到548g的3-氰基吡啶n-氧化物,收率为95%。
[0083]
2)2-氯-3-氰基吡啶的制备
[0084]
在3000ml的四口烧瓶中加入500g(4.16mol)3-氰基吡啶n-氧化物和2000g(13.04mol)三氯氧磷,搅拌,在冰水浴0-5℃下,滴加速度为400g/h滴加3摩尔当量的环己
胺,控制反应温度在0-5℃,控制反应体系的ph为9.5-10.5,滴加完毕后,升温至90℃下反应6-8h,检测到3-氰基吡啶n-氧化物小于1%;
[0085]
将反应液降至室温,在60℃下减压蒸馏,顶温45℃下回收得到1300g三氯氧磷。向剩余的浓缩液中加入1000g冰水稀释,搅拌状态下加入氢氧化钠溶液,调节ph至2-2.5,有大量褐色固体析出,继续在冰水浴下搅拌1h,抽滤干燥得到480g褐色固体,经1h nmr确认得到的固体产物为2-氯-3-氰基吡啶,2-氯-3-氰基吡啶摩尔收率为83%,2-氯-3-氰基吡啶的纯度为85.5%,通过高效液相色谱分析可知,剩余的14.5%杂质为6-氯-3-氰基吡啶。
[0086]
实施例4
[0087]
本实施例2-氯-3-氰基吡啶的制备方法如下:
[0088]
1)3-氰基吡啶n-氧化物的制备
[0089]
在3000ml的四口烧瓶中加入500g(4.80mol)3-氰基吡啶,1600g浓硫酸,升温至90℃至回流,回流状态下向反应体系中缓慢滴加400ml质量浓度为30%的双氧水,滴加完毕后继续回流反应2h,检测原料3-氰基吡啶已完全转化;
[0090]
将反应液降至室温下过滤得到固体产品,50℃下烘干称重,得到548g的3-氰基吡啶n-氧化物,收率为95%。
[0091]
2)2-氯-3-氰基吡啶的制备
[0092]
在3000ml的四口烧瓶中加入500g(4.16mol)3-氰基吡啶n-氧化物和2770g(18.06mol)三氯氧磷,搅拌,在冰水浴0-5℃下,滴加速度为每小时200g滴加3摩尔当量的环己胺,控制反应温度在0-5℃,控制反应体系的ph为9.5-10.5,滴加完毕后,升温至90℃下反应6-8h,检测到3-氰基吡啶n-氧化物小于1%;
[0093]
将反应液降至室温,在60℃下减压蒸馏,顶温45℃下回收得到2100g三氯氧磷。向剩余的浓缩液中加入1000g冰水稀释,搅拌状态下加入氢氧化钠溶液,调节ph至2-2.5,有大量褐色固体析出,继续在冰水浴下搅拌1h,抽滤干燥得到520g褐色固体,经1h nmr确认得到的固体产物为2-氯-3-氰基吡啶,2-氯-3-氰基吡啶摩尔收率为90%,2-氯-3-氰基吡啶的纯度为99.5%,通过高效液相色谱分析可知,剩余的0.5%杂质为6-氯-3-氰基吡啶。
[0094]
对比例1
[0095]
本对比例2-氯-3-氰基吡啶的制备方法如下:
[0096]
1)3-氰基吡啶n-氧化物的制备
[0097]
在3000ml的四口烧瓶中加入500g(4.80mol)3-氰基吡啶,1600g浓硫酸,升温至90℃至回流,回流状态下向反应体系中缓慢滴加400ml质量浓度为30%的双氧水,滴加完毕后继续回流反应2h,检测原料3-氰基吡啶已完全转化;
[0098]
将反应液降至室温下过滤得到固体产品,50℃下烘干称重,得到548g的3-氰基吡啶n-氧化物,收率为95%。
[0099]
2)2-氯-3-氰基吡啶的制备
[0100]
在3000ml的四口烧瓶中加入500g(4.16mol)3-氰基吡啶n-氧化物和2000g(13.04mol)三氯氧磷,搅拌,在冰水浴中以每小时200g的滴加速度滴加387.4g环己胺(4.16mol),控制反应温度在0-5℃以下,控制反应体系的ph为7.5-8.5,滴加完毕后,升温至90℃下反应6h,检测到3-氰基吡啶n-氧化物小于1%;
[0101]
将反应液降至室温,在60℃下减压蒸馏,顶温45℃下回收得到1300g三氯氧磷。向
剩余的浓缩液中加入1000g冰水稀释,搅拌状态下加入氢氧化钠溶液,调节ph至2-2.5,有大量褐色固体析出,继续在冰水浴下搅拌1h,抽滤干燥得到250g褐色固体,纯度为95%的2-氯-3-氰基吡啶,得到的2-氯-3-氰基吡啶的摩尔收率为43%。
[0102]
对比例2
[0103]
本对比例2-氯-3-氰基吡啶的制备方法如下:
[0104]
1)3-氰基吡啶n-氧化物的制备
[0105]
在3000ml的四口烧瓶中加入500g(4.80mol)3-氰基吡啶,1600g浓硫酸,升温至90℃至回流,回流状态下向反应体系中缓慢滴加400ml质量浓度为30%的双氧水,滴加完毕后继续回流反应2h,检测原料3-氰基吡啶已完全转化;
[0106]
将反应液降至室温下过滤得到固体产品,50℃下烘干称重,得到548g的3-氰基吡啶n-氧化物,收率为95%。
[0107]
2)2-氯-3-氰基吡啶的制备
[0108]
在3000ml的四口烧瓶中加入500g(4.16mol)3-氰基吡啶n-氧化物和2000g(13.04mol)三氯氧磷,搅拌,在冰水浴中以每小时200g的滴加速度滴加5.5摩尔当量的环己胺,控制滴加温度在0-5℃以下,控制反应体系的ph为12-13,滴加完毕后,升温至90℃下反应7h,检测到3-氰基吡啶n-氧化物小于1%;
[0109]
将反应液降至室温,在60℃下减压蒸馏,顶温45℃下回收得到1300g三氯氧磷。向剩余的浓缩液中加入1000g冰水稀释,搅拌状态下加入氢氧化钠溶液,调节ph至2-2.5,有大量褐色固体析出,继续在冰水浴下搅拌1h,抽滤干燥得到405g褐色固体,其中,2-氯-3-氰基吡啶摩尔收率为70%,经过高效液相色谱分析可知,褐色固体中含有80.5%的2-氯-3-氰基吡啶、19.5%的6-氯-3-氰基吡啶。
[0110]
以上各实施例仅用以说明本发明的技术方案,而非对其限制;尽管参照前述各实施例对本发明进行了详细的说明,本领域的普通技术人员应当理解:其依然可以对前述各实施例所记载的技术方案进行修改,或者对其中部分或者全部技术特征进行等同替换;而这些修改或者替换,并不使相应技术方案的本质脱离本发明各实施例技术方案的范围。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。