1.本发明属于基因表达数据分析领域,特别涉及一种基于基因表达的图神经网络数据集构建方法。
背景技术:
2.肿瘤疾病是全球主要公共健康问题之一,在肿瘤疾病治疗中生物标志物是一种最直接、快速有效的诊断手段,生物标志物的筛选与获得在疾病诊断、治疗以及疗效监测等多个方面发挥着重要作用,其同时也是药物开发的重要靶标。现今,发展创新性生物标志物的发现和鉴定方法,从而寻找和发现有价值的生物标志物,已经成为生物医学领域的重要研究热点之一。
3.随着高通量测序技术的快速发展,从基因组到表型各个层次的多分子水平数据越来越丰富,特别是基因表达谱数据。这为使用计算机方法发现肿瘤等复杂疾病的生物标志物提供了条件,但如何采用合适的算法处理大量的数据集是当前生物信息学与计算生物学研究面临的一个巨大挑战。近年来机器学习领域不断发展,深度学习是机器学习算法中的一个新兴技术,许多学者使用深度学习方法对生物标志物的发现方法进行了大量的研究,但在实际分析中还存在一些问题:
4.(1)大多是采取监督学习方法研究基因表达谱数据,但是生物信息分析获取标签数据的价格昂贵,且现有的标签数据难以满足需求;
5.(2)公共数据库收录的是样本与基因表达量的对应关系,缺乏不同样本之间的基因关联关系,相对于只关注差异表达的基因,讨论基因与基因之间的相互关系对于基因表达调控网络的研究更重要;
6.(3)图神经网络由于其处理非结构化数据时出色的能力,在社交网络、知识图、推荐系统以及生命科学等领域广泛应用,尤其是在图像数据中的应用,但是将公共数据库中大量的基因表达谱数据转换成图像显然不合理。
技术实现要素:
7.为解决上述问题,本发明提供了一种基于基因表达的图神经网络数据集构建方法,包括以下步骤:
8.s1.获取疾病的基因表达谱数据;
9.s2.从基因表达谱数据中提取rna数据,对rna数据进行处理生成rna表达矩阵;
10.s3.根据生成的rna表达矩阵进计算得到差异表达矩阵;
11.s4.通过差异表达矩阵,采用wgcna构建重叠拓扑矩阵;
12.s5.将重叠拓扑矩阵中的边信息与节点信息全部导出,对导出的信息进行编码;
13.s6.对编码后的信息进行筛选,采用筛选后的信息构建图神经网络数据集,根据图神经网络数据集识别生物标志物。
14.进一步的,步骤s1获取基因表达谱数据的过程为:
15.从公共数据库下载疾病样本数据;
16.获取人类参考基因组注释文件;
17.根据人类参考基因组注释文件整理疾病样本数据,得到基因表达谱数据。
18.进一步的,对rna数据进行处理的过程包括:
19.对rna数据进行id转换,将rna数据的ensemble id转换为symbol id;
20.设定低表达量阈值,将小于低表达量阈值的转换为symbol id后的rna数据过滤掉。
21.进一步的,采用wgcna构建重叠拓扑矩阵的过程,包括:
22.s11.根据皮尔森相关系数计算差异表达矩阵中基因之间的线性关系程度,根据线性关系生成关系矩阵;
23.s12.通过加权网络,将关系矩阵转换为邻接矩阵;
24.s13.根据转换生成的邻接矩阵构建重叠拓扑矩阵。
25.进一步的,关系矩阵表示为:s=[s
ij
]=[|cor(i,j)|];
[0026]
邻接矩阵表示为:a=[a
ij
]=[|cor(xi,xj)|
β
];
[0027]
重叠拓扑矩阵表示为:
[0028]
其中,i、j为基因,s
ij
表示基因i与基因j的相关性系数,s为关系矩阵,cor()为r包相关系数函数,β为网络符合无标度准则的软阈值,xi为基因i的基因表达量,a为邻接矩阵,u为遍历子,a
uj
为第u行第j列的基因表达量,a
iu
为第i行第u列的基因表达量,a
ij
第i行第j列的基因表达量;
[0029]
进一步的,对重叠拓扑矩阵中导出的边信息与节点信息进行编码后,采用scale函数进行标准化处理,表示为:
[0030][0031]
其中,n为公共数据库下载的疾病样本数量,x为疾病样本的基因表达量,z为标准分,标准分为求解过程即对数据的标准化过程。
[0032]
进一步的,对采用scale函数进行标准化处理后的信息进行筛选,包括:
[0033]
首先设定相关性阈值,根据相关性阈值对标准化处理后的边信息进行筛选;
[0034]
边信息筛选完成后,对节点信息进行筛选,满足最终节点数量是原始节点数量的90%以上。
[0035]
本发明的有益效果:
[0036]
本发明采取差异表达基因进行wgcna分析,关注基因与基因之间的关系,从而有利
于发现肿瘤的生物标志物;将重叠拓扑矩阵中的边信息与节点全部导出,针对导出的所有信息进行编码,再采用编过滤器和节点过滤器对编码的信息进行筛选,采用筛选后的信息构建图神经网络算法的输入数据集;使得能够采用图神经网络识别生物标志物,且筛选后构建的数据集使得识别效果更好,提升了识别效率和正确率。
附图说明
[0037]
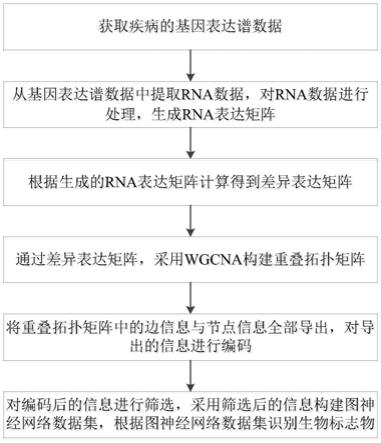
图1为本发明基于基因表达数据图神经网络数据集的构建方法流程图;
[0038]
图2为本发明差异基因筛选结果;
[0039]
图3为本发明软阈值筛选结果;
[0040]
图4为本发明模块特征基因可视化热图。
具体实施方式
[0041]
下面将结合本发明实施例中的附图,对本发明实施例中的技术方案进行清楚、完整地描述,显然,所描述的实施例仅仅是本发明一部分实施例,而不是全部的实施例。基于本发明中的实施例,本领域普通技术人员在没有做出创造性劳动前提下所获得的所有其他实施例,都属于本发明保护的范围。
[0042]
一种基于基因表达的图神经网络数据集构建方法,如图1所示,包括以下步骤:
[0043]
s1.获取疾病的基因表达谱数据;
[0044]
s2.从基因表达谱数据中提取rna数据,对rna数据进行处理,生成rna表达矩阵;
[0045]
s3.根据生成的rna表达矩阵计算得到差异表达矩阵;
[0046]
s4.通过差异表达矩阵,采用wgcna构建重叠拓扑矩阵;
[0047]
s5.将重叠拓扑矩阵中的边信息与节点信息全部导出,对导出的信息进行编码;
[0048]
s6.对编码后的信息进行筛选,采用筛选后的信息构建图神经网络数据集,根据图神经网络数据集识别生物标志物。
[0049]
在一实施例中,使用tcga数据库中的脑胶质瘤基因表达谱数据进行分析,将下载得到的脑胶质瘤基因表达谱数据通过人类crch38参考基因组注释文件注释,提取脑胶质瘤基因表达谱数据中的rna数据,将rna数据进行id转换,即将rna数据的ensemble id转换为symbol id,ensembleid是数据库使用的id标识符,而本发明分析的是基因表达量之间的关系,所以需要转换成symbolid,symbolid就是对应的基因名称;
[0050]
具体地,rna数据包括mrna和lncrna数据;
[0051]
设定低表达量阈值,id转换过后,根据低表达量阈值筛选去除低表达量的rna数据获得rna表达矩阵;本实施例中,低表达量阈值为40,即该基因在95%的疾病样本中均不表达,将一个在所有疾病样本中表达总和小于40的基因过滤掉,这样做的目的是去除低表达量的基因对总体基因表达量的影响。
[0052]
标准化是将数据按比例缩放,使之落入一个比较小的特定范围,
[0053]
优选地,对获取的rna表达矩阵进行标准化处理,这里的标准化处理主要是用r语言的分位数标准化函数normalizebetweenarrays()进行的,标准化处理完成后进行log变换得到标准化表达矩阵,对标准化表达矩阵进行分组,将恶性脑胶质瘤作为参考组,低级脑胶质瘤作为对照组进行差异分析,差异基因筛选结果如图2所示,图2为差异分析火山图,横
坐标为log2(fold change),表示一个基因在两样本中表达量差异倍数的对数值,其绝对值越大,说明表达量在两样本间的表达量倍数差异越大,纵坐标为-log10(p值),表示fdr的负对数值,其值越大,表明差异表达越显著,筛选得到的差异表达基因越可靠,差异分析火山图中的每一个点表示一个基因;
[0054]
优选地,本实施例中的差异分析是使用r语言的limma包和edger包完成的,包括:
[0055]
101、使用calcnormfactors()函数计算表达矩阵的归一化因子以对齐计数矩阵的列;
[0056]
102、使用voom()函数转换表达矩阵数据为线性建模做准备;
[0057]
103、使用lmfit()函数为每个基因拟合线性模型;
[0058]
104、使用contrasts.fit()函数根据构造的对比矩阵计算给定对比组的估计系数和标准误差;
[0059]
105、使用ebayes()函数进行差分表达的贝叶斯统计。
[0060]
在一实施例中,采用wgcna分析差异表达矩阵并构建重叠拓扑矩阵,其主要过程包括:
[0061]
s11.根据皮尔森相关系数计算差异表达矩阵中基因之间的线性关系程度,根据线性关系生成关系矩阵;
[0062]
具体地,关系矩阵表示为:s=[s
ij
]=[|cor(i,j)|];
[0063]
s12.通过加权网络,将关系矩阵转换为邻接矩阵;由于未加权网络的点只存在0和1的关系,所能翻译的信息较少,不能反应点和点之间关系的强弱,故在本实施例中,采用加权网络,通过生成包含不同阈值的数列,遍历给定的阈值,找到符合无标度网络准则的软阈值,如图3所示,左图中的线表示主观选择的无标度拟合指数取值,如图为0.9,将关系矩阵转换成邻接矩阵;
[0064]
具体地,邻接矩阵表示为:a=[a
ij
]=[|cor(xi,xj)|
β
];
[0065]
s13.根据转换生成的邻接矩阵构建重叠拓扑矩阵;
[0066]
具体地,重叠拓扑矩阵表示为:
[0067][0068]
其中,i、j为基因,s
ij
表示基因i与基因j的相关性系数,s为关系矩阵,cor()为r包相关系数函数,β为网络符合无标度准则的软阈值,xi为基因i的基因表达量,a为邻接矩阵,u为遍历子,a
uj
为第u行第j列的基因表达量,a
iu
为第i行第u列的基因表达量,a
ij
第i行第j列的基因表达量,tom为重叠拓扑矩阵;基因i与基因j在网络中的连接情况越相似,tom
ij
值越大,基因i和基因j就越可能在同一表达模块,即同一分类组别中,图4为模块特征基因可视化图。
[0069]
在一实施例中,采用wgcna分析差异表达矩阵并构建重叠拓扑矩阵,将重叠拓扑矩阵中的边信息与节点信息全部导出,对导出的信息进行编码后,采用scale函数进行标准化处理,表示为:
[0070][0071]
其中,n为公共数据库下载的疾病样本数量,x为疾病样本的基因表达量,z为标准分。
[0072]
优选地,对采用scale函数进行标准化处理后的信息进行筛选,包括:
[0073]
首先对标准化处理后的边信息进行筛选:设定相关性阈值,若边的相关性大于相关性阈值,则保留此边,若边的相关性小于相关性阈值,则去除此边;
[0074]
本实施例中,根据scale函数标准化后的权重信息,设定相关性阈值为0.5;
[0075]
边信息筛选完成后,对节点信息进行筛选,满足最终节点数量是原始节点数量的90%以上。
[0076]
其中,相关性为wgcna分析得到的tom矩阵中包含的节点与节点之间的权重值。
[0077]
由于采用wgcna得到的tom矩阵导出的边和节点的信息为完全图,完全图对于后续采用图神经网络算法对数据集进行分类预测是没有用的,并且完全图在基因表达中也不符合实际,因此需要通过对边和节点的筛选来修剪完全图,从而获取基因与基因之间的相关性。筛选的两个过滤器分别是对边的筛选和节点的筛选。
[0078]
边信息筛选过滤器为:至少每一个节点对应一条边,且边的平均度为最优;
[0079]
节点信息筛选过滤器为:节点比例大于90%;
[0080]
具体地,对边的筛选所要遵循的是:在这个图中每个节点至少都有一条边和另一个节点进行连接,由于边与边的连接相关性就代表了基因与基因之间关系的相关性,所以留下的边越少就代表这些边所连接的节点越相关,边的平均度的计算是图中全部边的数量除以节点的数量,度越小就代表图中的节点越相关。
[0081]
采用筛选后的信息构建图神经网络数据集,如表1所示,根据上述过程,构建7个图神经网络数据集,如表2所示,使用python语言编码将7个图神经网络数据集转换成数字;在本实施例中,共使用7个图嵌入算法(hope、gf、grarep、sdne、deepwalk、line、node2vec)对所产生的图神经网络数据集进行评估,评估指标是micro(微平均)和macro(宏平均),并与图嵌入公共数据集进行比较,根据图神经网络数据集的评估结果识别生物标志物。
[0082]
表1图神经网络数据集
[0083][0084]
表2图神经网络数据集转换基础编码
[0085][0086]
尽管已经示出和描述了本发明的实施例,对于本领域的普通技术人员而言,可以理解在不脱离本发明的原理和精神的情况下可以对这些实施例进行多种变化、修改、替换和变型,本发明的范围由所附权利要求及其等同物限定。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。