1.本发明属于有机合成技术领域,具体涉及一种二苯基甲烷类化合物及其制备方法。
背景技术:
2.随着化学试剂种类的丰富,不仅推动医药技术的快速发展,材料化学领域也是日新月异。二苯基甲烷类化合物也逐渐在化学材料领域有着广泛的应用。测试显示二苯基甲烷类化合物具有良好的化学选择性,耐受各种反应性官能团,由其制成的化学材料普遍具有耐高温、耐腐蚀和优越的机械性能。且具有原料易得、反应高效、低毒及方便合成等优点。
3.对于二苯基甲烷类化合物的新型合成方法的开发也在持续优化中。目前,二苯基甲烷类化合物的合成方法主要是金属催化的苯甲醇与芳香族溴化物的交叉亲电偶联反应得到。其反应过程复杂,副反应过多。而对于羟基还原的方法普遍是参考黄鸣龙反应用水合肼高温还原,操作过程较为危险。其他的金属催化还原较难反应完全,且应用范围较窄。
技术实现要素:
4.针对现有技术中的不足,本发明的目的是提供一种二苯基甲烷类化合物及其制备方法。
5.为达到上述目的,本发明的解决方案是:
6.第一方面,本发明提供了一种二苯基甲烷类化合物的制备方法,其包括如下步骤:
7.(1)、碘、二苯基甲醇类化合物和乙酸在氮气保护下搅拌均匀,得到第一混合液;在第一混合液中加入还原剂,得到第二混合液;
8.(2)、将第二混合液加热,然后进行搅拌,薄层色谱法检测反应完毕,经过萃取,洗涤,干燥后浓缩得到二苯基甲烷类化合物。
9.作为本发明的优选实施例,步骤(1)中,二苯基甲醇类化合物中的官能团选自氯、溴、甲基、甲氧基或氢中的一种以上。
10.作为本发明的优选实施例,步骤(1)中,搅拌的温度为20-25℃,搅拌的时间为3-10min。
11.作为本发明的优选实施例,步骤(1)中,还原剂为次磷酸(h3po2)。
12.作为本发明的优选实施例,步骤(2)中,加热的温度为40-70℃,加热的时间为1.5-2.5h。
13.作为本发明的优选实施例,步骤(2)中,搅拌的时间为2-8h。
14.作为本发明的优选实施例,步骤(2)中,干燥为室温干燥时,干燥的温度为20-25℃,干燥的时间为22-26h。
15.作为本发明的优选实施例,步骤(2)中,干燥为烘箱干燥时,干燥的温度为40-50℃,干燥的时间为3-5h。
16.第二方面,本发明提供了一种二苯基甲烷类化合物,其由上述的制备方法得到。
17.具体地,二苯基甲烷类化合物的结构通式为:
[0018][0019]
其中,r1选自氯、溴、甲基或甲氧基中的一种以上,r2选自氯、溴、甲基、甲氧基或氢中的一种以上。
[0020]
由于采用上述方案,本发明的有益效果是:
[0021]
第一、现有技术中使用水合肼进行的金属还原反应往往需要高温(超过100℃),而本发明的制备方法在不超过70℃,8h之内就能高效反应完全,从而扩大了使用范围;另外,本发明的反应能安全、可靠、温和地由二苯甲醇化合物生成二苯甲烷类化合物反应,且无危险气体和其他危险产物产生,杂质易除去,反应干净,得到产物纯度高无需柱层析纯化,且收率较高。
[0022]
第二、本本发明的制备方法操作简单,加入原料简单,反应原理单一。
附图说明
[0023]
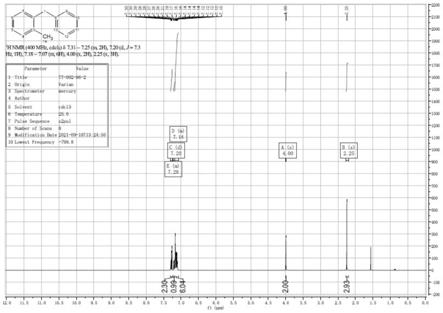
图1为本发明的实施例1中2-甲基二苯基甲烷的1h-nmr图。
[0024]
图2为本发明的实施例1中2-甲基二苯基甲烷的gcms图。
[0025]
图3为本发明的实施例2中4-甲氧基二苯基甲烷的1h-nmr图。
[0026]
图4为本发明的实施例2中4-甲氧基二苯基甲烷的gcms图。
[0027]
图5为本发明的实施例3中4,4-二甲基二苯基甲烷的1h-nmr图。
[0028]
图6为本发明的实施例3中4,4-二甲基二苯基甲烷的gcms图。
[0029]
图7为本发明的实施例4中(4-溴苯基)(苯基)甲烷的1h-nmr图。
[0030]
图8为本发明的实施例4中(4-溴苯基)(苯基)甲烷gcms图。
[0031]
图9为本发明的实施例5中4,4-二甲氧基二苯基甲烷的1h-nmr图。
[0032]
图10为本发明的实施例5中4,4-二甲氧基二苯基甲烷的gcms图。
[0033]
图11为本发明的实施例6中4,4-二氯二苯基甲烷的1h-nmr图。
[0034]
图12为本发明的实施例6中4,4-二氯二苯基甲烷的lcms图。
具体实施方式
[0035]
本发明提供了一种二苯基甲烷类化合物及其制备方法。
[0036]
《二苯基甲烷类化合物的制备方法》
[0037]
本发明的二苯基甲烷类化合物的制备方法包括如下步骤:
[0038]
(1)、碘(i2,1eq)、(1mmol)二苯基甲醇类化合物和(6ml)乙酸(acoh)在装有冷凝器的烧瓶中在氮气保护下搅拌均匀,得到第一混合液,即为黑色溶液;然后在第一混合液中加入还原剂(h3po2)(50%aq.,0.5ml;5mmol),溶液颜色逐渐变淡,得到第二混合液,即最后为黄棕色溶液;
[0039]
(2)、滴完并将第二混合液加热,然后进行搅拌,此时溶液淡黄澄清,薄层色谱法(tlc)检测反应完毕(tlc:pe/ea=10)。加水稀释,此时溶液变浑浊,为白色悬浊液。用10ml石油醚(pe)萃取,水相用3ml的pe洗涤,合并的有机相用饱和食盐水清洗完,在无水na2so4上室温干燥或放置烘箱干燥后浓缩得到无色液体。收率为80-95%,纯度为95-98%。
[0040]
反应方程式为:
[0041][0042]
其中,r1选自cl、br、ch3或och3中的一种以上,r2选自cl、br、ch3、och3或h中的一种以上。
[0043]
在步骤(1)中,碘作为还原剂和催化剂参与反应,乙酸作为溶剂和还原剂次磷酸组合形成高效还原剂。
[0044]
在步骤(1)中,搅拌的温度可以为20-25℃,优选为25℃;搅拌的时间可以为3-10min,优选为5min。
[0045]
在步骤(2)中,加热的温度可以为40-70℃,优选为50-70℃,更优选为60℃;加热的时间可以为1.5-2.5h,优选为2h,一般加热搅拌2h后可进行tlc检测,如原料反应完毕立即停止反应。
[0046]
在步骤(2)中,搅拌的时间可以为1-8h,优选为1-5h,更优选为2h。
[0047]
在步骤(2)中,干燥为室温干燥时,干燥的温度可以为20-25℃,优选为25℃;干燥的时间可以为22-26h,优选为24h。
[0048]
在步骤(2)中,干燥为烘箱干燥时,干燥的温度可以为40-50℃,优选为45℃;干燥的时间可以为3-5h,优选为4h。
[0049]
《二苯基甲烷类化合物》
[0050]
本发明的二苯基甲烷类化合物由上述的制备方法得到。
[0051]
具体地,二苯基甲烷类化合物的结构通式为:
[0052][0053]
其中,r1选自cl、br、ch3或och3中的一种以上,r2选自cl、br、ch3、och3或h中的一种以上。
[0054]
以下结合实施例对本发明作进一步的说明。
[0055]
实施例1:
[0056]
本实施例的2-甲基二苯基甲烷的制备方法包括如下步骤:
[0057]
(1)、碘(7.5g,30mmol)、2-甲基二苯基甲醇(5.99g,30mmol)和乙酸(190ml)在装有冷凝器的烧瓶中在氮气保护下搅拌均匀,得到第一混合液,即为黑色溶液;其中,搅拌的温度为25℃,搅拌的时间为5min。然后在第一混合液中加入次磷酸(50%aq.,15ml;145mmol),溶液颜色逐渐变淡,得到第二混合液,即最后为黄棕色溶液。
[0058]
(2)、滴完并将第二混合液加热至60℃,加热的时间为2h,然后搅拌2h,此时溶液淡黄澄清,tlc检测反应完毕。加入200ml水稀释,此时溶液变浑浊,为白色悬浊液。用pe(200ml)萃取,水相用50ml的pe洗涤,合并的有机相用饱和食盐水清洗完,在无水na2so4上室温25℃干燥24h后浓缩得到5.4g无色液体。纯度为98%以上,收率为96%。如图1和图2所示。
[0059]
其中,反应方程式为:
[0060][0061]
表1各试剂的参数
[0062]
试剂分子式分子量投料量投料比2-甲基二苯基甲醇c
14h14
o198.275.99g1.0eq.次磷酸h3po26615ml4.8eq.乙酸c2h4o260.05190ml/碘i2253.87.5g1.0eq
[0063]
实施例2:
[0064]
本实施例的4-甲氧基二苯基甲烷的制备方法包括如下步骤:
[0065]
(1)、碘(7.5g,30mmol)、4-甲氧基二苯基甲醇(6.5g,30mmol)和乙酸(190ml)在装有冷凝器的烧瓶中在氮气保护下搅拌均匀,得到第一混合液,即为黑色溶液;其中,搅拌的温度为20℃,搅拌的时间为3min。然后在第一混合液中加入次磷酸(50%aq.,15ml;145mmol),溶液颜色逐渐变淡,得到第二混合液,即最后为黄棕色溶液。
[0066]
(2)、滴完并将第二混合液加热至50℃,加热的时间为2.5h,然后搅拌1h,此时溶液淡黄澄清,tlc检测反应完毕。加入200ml水稀释,此时溶液变浑浊,为白色悬浊液。用pe(200ml)萃取,水相用50ml的pe洗涤,合并的有机相用饱和食盐水清洗完,在无水na2so4上烘箱45℃干燥4h干燥后浓缩得到5.5g淡黄色液体。纯度为98%以上,收率为90%。如图3和图4所示。
[0067]
其中,反应方程式为:
[0068][0069]
表2各试剂的参数
[0070][0071]
实施例3:
[0072]
本实施例的4,4-二甲基二苯基甲烷的制备方法包括如下步骤:
[0073]
(1)、4,4-二甲基二苯基甲醇(10g)、碘(11.96g)和乙酸(300ml)在装有冷凝器的烧瓶中在氮气保护下搅拌均匀,得到第一混合液,即为黑色溶液;其中,搅拌的温度为25℃,搅拌的时间为3min。然后在第一混合液中加入次磷酸(50%aq.,23.9ml),溶液颜色逐渐变淡,得到第二混合液,即最后为黄棕色溶液。
[0074]
(2)、滴完并将第二混合液加热至40℃,加热的时间为1.5h,然后搅拌4h,此时溶液淡黄澄清,tlc检测反应完毕。加入300ml水稀释,此时溶液变浑浊,为白色悬浊液。用pe(300ml)萃取,水相用50ml的pe洗涤,合并的有机相用饱和食盐水清洗完,在无水na2so4上室温25℃干燥22h后浓缩得到约9.1g黄色油状的化合物。纯度为98%以上,收率为90%。如图5和图6所示。
[0075]
其中,反应方程式为:
[0076][0077]
表3各试剂的参数
[0078][0079]
实施例4:
[0080]
本实施例的(4-溴苯基)(苯基)甲烷的制备方法包括如下步骤:
[0081]
(1)、(4-溴苯基)(苯基)甲醇(10g)、碘(9.65g)和乙酸(240ml)在装有冷凝器的烧瓶中在氮气保护下搅拌均匀,得到第一混合液,即为黑色溶液;其中,搅拌的温度为23℃,搅拌的时间为8min。然后在第一混合液中加入次磷酸(50%aq.,19.3ml),溶液颜色逐渐变淡,得到第二混合液,即最后为黄色溶液。
[0082]
(2)、滴完并将第二混合液加热至70℃,加热的时间为2h,然后搅拌8h,此时溶液淡黄澄清,tlc检测反应完毕。加入200ml水稀释,此时溶液变浑浊,为白色悬浊液。用pe(200ml)萃取,水相用50ml的pe洗涤,合并的有机相用饱和食盐水清洗完在无水na2so4上烘箱40℃干燥3h后浓缩得到约9.6g淡黄色油状化合物。纯度为97%以上,收率为97%。如图7和图8所示。
[0083]
其中,反应方程式为:
[0084][0085]
表4各试剂的参数
[0086][0087]
实施例5:
[0088]
本实施例的4,4-二甲氧基二苯基甲烷的制备方法包括如下步骤:
[0089]
(1)、4,4-二甲氧基二苯基甲醇(15g)、碘(15.6g)和乙酸(390ml)在装有冷凝器的烧瓶中在氮气保护下搅拌均匀,得到第一混合液,即为黑色溶液;其中,搅拌的温度为25℃,搅拌的时间为5min。然后在第一混合液中加入次磷酸(50%aq.,31.2ml),溶液颜色逐渐变淡,得到第二混合液,即最后为黄色溶液。
[0090]
(2)、滴完并将第二混合液加热至50℃,加热的时间为2h,然后搅拌3h,此时溶液淡黄澄清,tlc检测反应完毕。加入300ml水稀释,此时溶液变浑浊,为白色悬浊液。用pe(350ml)萃取,水相用10ml的pe洗涤,合并的有机相用饱和食盐水清洗完,在无水na2so4上烘箱50℃干燥5h后浓缩得到约13.5g无色油状的化合物。纯度为98%以上,收率为90%。如图9和图10所示。
[0091]
其中,反应方程式为:
[0092][0093]
表5各试剂的参数
[0094][0095]
实施例6:
[0096]
本实施例的4,4-二氯二苯基甲烷的制备方法包括如下步骤:
[0097]
(1)、4,4-二氯二苯基甲醇(17g)、碘(17.05g)和乙酸(430ml)在装有冷凝器的烧瓶中在氮气保护下搅拌均匀,得到第一混合液,即为黑色溶液;其中,搅拌的温度为25℃,搅拌的时间为5min。然后在第一混合液中加入次磷酸(50%aq.,34.1ml),溶液颜色逐渐变淡,得到第二混合液,即最后为黄棕色溶液。
[0098]
(2)、滴完并将第二混合液加热至70℃,加热的时间为2h,然后搅拌5h,此时溶液淡黄澄清,tlc检测反应完毕。加入400ml水稀释,此时溶液变浑浊,为白色悬浊液。用pe
(300ml)萃取,水相用100ml的pe洗涤,合并的有机相用饱和食盐水清洗完,在无水na2so4上室温25℃干燥26h后浓缩得到约20g无色油状的化合物,加入20ml的pe在0℃打浆,有白色晶体析出,15min后将固体滤出晾干得到15.6g双(4-氯苯基)甲烷(即4,4-二氯二苯基甲烷),为白色晶体。纯度为98%以上,收率为92%。如图11和图12所示。
[0099]
其中,反应方程式为:
[0100][0101]
表6各试剂的参数
[0102][0103]
上述对实施例的描述是为了便于该技术领域的普通技术人员能理解和使用本发明。熟悉本领域技术人员显然可以容易的对这些实施例做出各种修改,并把在此说明的一般原理应用到其他实施例中,而不必经过创造性的劳动。因此,本发明不限于上述实施例。本领域技术人员根据本发明的原理,不脱离本发明的范畴所做出的改进和修改都应该在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。