1.本发明涉及分析化学技术领域,具体涉及一种针对植物源性食品中砜吡草唑残留量的气相色谱-三重四极杆质谱检测方法。
背景技术:
2.砜吡草唑(pyroxasulfone,代号:kih-485)是日本组合化学公司开发的新型广谱、高活性的苗前土壤处理除草剂。该除草剂属于异噁唑类除草剂,是植物体内超长链脂肪酸(vlcfas)生物合成的潜在抑制剂。砜吡草唑的应用作物较为广泛,可以安全地用于玉米、棉花、花生、小麦、向日葵、高粱等作物;可有效防除狗尾草属、马唐属、稗属等禾本科杂草以及苋属、曼陀罗属、茄属、苘麻属等阔叶杂草。砜吡草唑在澳大利亚等国家被认为是防除硬直黑麦草等抗药性杂草的最佳药剂。砜吡草唑具有杀草谱广、活性高、用量低、安全性好等优良特点,使其受到越来越广泛的关注,近年来已经在澳大利亚、美国、加拿大等国家登记在小麦、玉米、大豆、棉花等作物田封闭除草,其中澳大利亚制定了玉米、谷类、豆类种子、大豆(干)和葵花籽等植物源食品中的残留限量,残留限量值低至0.01mg/kg,目前我国也逐步将砜吡草唑作为麦田最新的土壤处理剂在进行登记和推广。
3.针对砜吡草唑的研究主要集中在除草活性、环境行为和安全性评价方面,对于砜吡草唑的残留分析方法,国内外相关报道较少。吴文铸等使用高效液相色谱法建立了土壤与水中砜吡草唑残留检测方法,而对于砜吡草唑在粮谷等植物源食品中的分析方法尚未见报道。因此,开发植物源食品中的砜吡草唑残留检测分析方法,可弥补我国该项目没有检测方法的空白;对评估和管理该化合物对人类健康和环境安全的影响,防止贸易输入输出风险具有极其重要的作用。
技术实现要素:
4.本发明旨在针对现有技术的技术缺陷,提供一种针对植物源性食品中砜吡草唑残留量的气相色谱-三重四极杆质谱检测方法,以解决砜吡草唑在粮谷等植物源食品中的分析方法尚未建立的技术问题。
5.为实现以上技术目的,本发明采用以下技术方案:
6.针对植物源性食品中砜吡草唑残留量的气相色谱-三重四极杆质谱检测方法,包括以下步骤:取待测植物源性样品粉碎后充分混匀;称取5g粉碎后的试样于50ml聚丙烯离心管中,加入3g氯化钠、10ml乙酸乙酯及陶瓷均质子,涡旋振荡提取5min;4000r/min离心3min;吸取1.5ml上清液于2ml聚丙烯离心管中,加入20mgpsa、20mg c
18
和50mg无水硫酸镁;涡旋混合1min,12000r/min离心3min,取上清液过0.22μm有机滤膜;而后以如下仪器条件执行检测:
7.db-17ms毛细管色谱柱;柱箱升温程序:70℃保持0min,然后以20℃/min升温至230℃,保持0min;最后以30℃/min升温至310℃,保持1min;载气:氦气,纯度≥99.999%,恒流
模式,流量为1.0ml/min;进样口温度:280℃;进样量:1l;进样方式:不分流进样;电子轰击源:70ev;离子源温度:230℃;传输线温度:280℃;溶剂延迟:3min;多反应监测模式。
8.作为优选,在检测过程中保留时间为8.71min,定量离子对及其碰撞能量分别为228.6/179.1、15ev,定性离子对及其碰撞能量分别为178.6/159.0、15ev,回归方程为y=3038.846525x-1470.727423,相关系数为0.9997,线性范围为0.001-0.05mg/kg,检测限为0.001mg/kg。
9.作为优选,待测植物源性样品选自以下成分的其中一种或其中若干种:稻谷、小麦、高粱、玉米、大豆、花生、甘薯、马铃薯、葵花籽。
10.作为优选,所述db-17ms毛细管色谱柱的规格为30m
×
0.25mm
×
0.25μm。
11.作为优选,还包括以下步骤:配制若干已知其中砜吡草唑浓度的基质混合标准工作溶液,并绘制标准曲线。
12.作为优选,所述基质混合标准工作溶液是由以下方法配制的:吸取100mg/l标准储备液,用乙酸乙酯配制成1mg/l的混合标准溶液,0℃~4℃避光保存,有效期1个月;吸取混合标准溶液,用空白样品提取液配成浓度为0.5μg/l、2μg/l、5μg/l、10μg/l和25μg/l的基质混合标准工作溶液。
13.本发明提供了一种针对植物源性食品中砜吡草唑残留量的气相色谱-三重四极杆质谱检测方法。该技术方案将稻谷、小麦、高粱、玉米、大豆、花生、甘薯、马铃薯和葵花籽中的砜吡草唑残留用乙酸乙酯提取,经乙二胺-n-丙基硅烷化硅胶、十八烷基键合硅胶和无水硫酸镁去除杂质,离心过滤后经气相色谱-三重四极杆质谱仪检测,基质匹配外标法定量。实验对提取溶剂、提取方式、净化方式和仪器条件进行了优化,考察了基质效应的影响,确定出最优的前处理方法和仪器条件。在优化实验条件下,目标化合物在0.001-0.05mg/kg范围内的线性关系良好,相关系数大于0.99。空白样品在低、中、高4个添加水平下的平均回收率为83.8%-104.3%,相对标准偏差(n=6)为2.6%-6.7%,方法定量限为0.001mg/kg。该方法操作简单、快速、灵敏,能满足植物源食品中砜吡草唑的检测要求。本发明通过优化色谱条件,考察提取溶剂类型、净化剂、基质效应对回收率的影响,建立了砜吡草唑的quechers-气相色谱三重四极杆质谱检测方法,为植物源性产品中砜吡草唑残留的检测提供了坚实的技术保障。
附图说明
14.图1是砜吡草唑的总离子流图。
15.图2是砜吡草唑的提取离子mrm色谱图。
16.图3是提取溶液类型-平均回收率关系图。
17.图4是不同吸附剂对加标空白样品的提取回收率的影响图。
18.图5是不同样品中异硫氰酸甲酯和棉隆的基质效应me值。
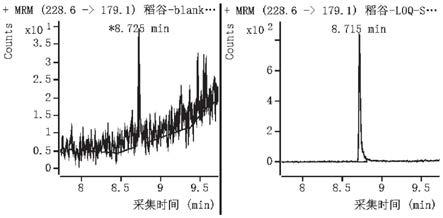
19.图6是稻谷空白样品及定量限(0.001mg/kg)加标样品的mrm色谱图。
20.图7是小麦空白样品及定量限(0.001mg/kg)加标样品的mrm色谱图。
21.图8是高粱空白样品及定量限(0.001mg/kg)加标样品的mrm色谱图。
22.图9是玉米空白样品及定量限(0.001mg/kg)加标样品的mrm色谱图。
23.图10是大豆空白样品及定量限(0.001mg/kg)加标样品的mrm色谱图。
24.图11是花生空白样品及定量限(0.001mg/kg)加标样品的mrm色谱图。
25.图12是甘薯空白样品及定量限(0.001mg/kg)加标样品的mrm色谱图。
26.图13是马铃薯空白样品及定量限(0.001mg/kg)加标样品的mrm色谱图。
27.图14是葵花籽空白样品及定量限(0.001mg/kg)加标样品的mrm色谱图。
具体实施方式
28.以下将对本发明的具体实施方式进行详细描述。为了避免过多不必要的细节,在以下实施例中对属于公知的结构或功能将不进行详细描述。以下实施例中所使用的近似性语言可用于定量表述,表明在不改变基本功能的情况下可允许数量有一定的变动。除有定义外,以下实施例中所用的技术和科学术语具有与本发明所属领域技术人员普遍理解的相同含义。
29.1、试剂和仪器
30.1.1试剂
31.无水硫酸镁(分析纯,使用前于650℃下灼烧4h,备用)和氯化钠购于国药集团化学试剂有限公司;正己烷、丙酮、乙酸乙酯为色谱纯,购于美国tedia公司。乙二胺-n-丙基硅烷化硅胶(psa:40μm~60μm)和十八烷基键合硅胶(c
18
:40μm~60μm)购于上海安谱实验科技股份有限公司。微孔滤膜:0.22μm,有机相型,陶瓷均质子:2cm(长)
×
1cm(外径)。砜吡草唑(c
12h14
f5n3o4s,cas no.447399-55-5),100μg/ml,不确定度
±
3%(k=2)购于bepure公司。气相色谱-三重四极杆质谱联用仪(美国安捷伦7000b):配备电子轰击源(ei)、分析天平:感量0.01g和0.0001g(德国赛多利斯)、粉碎机(上海嘉定粮油仪器有限公司)、涡旋振荡器(美国talboys公司)、离心机:转速不低于4000r/min(无锡市瑞江分析仪器有限公司)、离心机:转速不低于12000r/min(湖南湘仪离心机有限公司)。
32.1.2实验方法
33.1.2.1标准溶液配制标准溶液:吸取适量的100mg/l标准储备液,用乙酸乙酯配制成1mg/l的混合标准储备液,0℃~4℃避光保存,有效期1个月。
34.基质混合标准工作溶液:吸取适量的混合标准溶液,用空白样品提取液配成浓度为0.5μg/l、2μg/l、5μg/l、10μg/l和25μg/l的基质混合标准工作溶液,基质混合标准工作溶液应现配现用。
35.1.2.2 quechers前处理
36.稻谷、小麦、玉米和花生等植物源性样品粉碎后充分混匀。称取5g(精确至0.01g)粉碎后的试样于50ml聚丙烯离心管中,加入3g氯化钠、10ml乙酸乙酯及一颗陶瓷均质子,涡旋振荡提取5min;n;4000r/min离心3min。准确吸取1.5ml上清液于2ml聚丙烯离心管中,加入20mgpsa、20mg c
18
和50mg无水硫酸镁。涡旋混合1min,12000r/min离心3min,取上清液过0.22μm有机滤膜,用于测定。
37.1.2.3仪器条件
38.db-17ms毛细管色谱柱(30m
×
0.25mm
×
0.25μm)柱或相当者;柱箱升温程序:70℃保持0min,然后以20℃/min升温至230℃,保持0min;最后以30℃/min升温至310℃,保持1min;载气:氦气,纯度≥99.999%,恒流模式,流量为1.0ml/min;进样口温度:280℃;进样量:1μl;进样方式:不分流进样;电子轰击源:70ev;离子源温度:230℃;传输线温度:280℃;
溶剂延迟:3min;多反应监测模式:选择一对定量离子、一对定性离子,其保留时间、定量离子对、定性离子对和碰撞能量,参见表1。
39.表1砜吡草唑的保留时间、定量离子对、定性离子对、碰撞能量、回归方程、相关系数、线性范围和定量限
[0040][0041]
2结果与讨论
[0042]
2.1仪器条件
[0043]
2.1.1仪器的选择
[0044]
本研究旨在建立对砜吡草唑进行检测并确证的检测方法,在选择检测仪器时放弃选择使用气相色谱仪和液相色谱仪,仅选择尝试定性能力更强的高效液相色谱-串联质谱仪和三重四极杆气质联用仪。首先按照高效液相色谱-串联质谱仪方法开发程序对棉隆和异硫氰酸甲酯进行母离子扫描和子离子扫描,实验发现砜吡草唑的母离子391.2响应较好,但优化后上柱分析峰型一般响应较差,检测限较高无法满足相关法规要求;最终放弃使用高效液相色谱-串联质谱法对砜吡草唑进行检测,而三重四极杆气质联用仪能得到峰型良好,方法灵敏度高,最终确定使用三重四极杆气质联用仪用于植物源性食品中砜吡草唑残留量的测定。
[0045]
2.1.2质谱条件优化
[0046]
选择5mg/l的砜吡草唑标准溶液进行ms1全扫描,选择响应较高的离子作为母离子,确定砜吡草唑的母离子228.6和178.6,再对相应母离子进行产物离子扫描,选择信号最好的两组作为mrm定量和定性离子对。0.01mg/l基质标准溶液中砜吡草唑的总离子流图、砜吡草唑提取离子mrm色谱图见图1和图2。
[0047]
2.2前处理方法的优化
[0048]
2.2.1提取方式和提取溶剂的选择
[0049]
实验选择正己烷、正己烷-丙酮(9∶1,v/v)、正己烷-丙酮(2∶1,v/v)、正己烷-丙酮(7∶3,v/v)和乙酸乙酯作为提取溶剂,将添加0.01mg/kg砜吡草唑的小麦样品按照1.2的实验方法处理上机,结果平行测定3次,得到提取溶液类型-平均回收率关系图如图3所示,结果表明五种提取溶剂对于砜吡草唑的提取效率均较好,从提取液颜色看,含有丙酮的混合溶剂提取液颜色稍深且随丙酮含量增加而增加,从绿色环保、降低成本及操作简便的方面考虑,最终选择乙酸乙酯作为提取溶剂。
[0050]
2.2.2净化方式的选择
[0051]
乙二胺-n-丙基硅烷化硅胶(psa)是弱阴离子交换吸附剂,可以有效去除样品中的有机酸、极性色素、脂肪酸、糖类以及其他能形成氢键的成分;十八烷基硅烷键合硅胶(c
18
)可去除如挥发油、萜类、木质素、脂类、类胡萝卜素等非极性化合物,无水硫酸镁可以去除样液中的水分。quechers法进行粮谷和油料样品的前处理净化时通常使用psa、c18和无水硫
酸镁的组合用以去除样液中的杂质和水分。为使实验过程尽量简便快速,前处理过程采用一次提取不浓缩,quechers净化的方式进行。实验考察了psa、c
18
和无水mgso4三种物质对砜吡草唑标准溶液进行吸附后的回收率。称取50mg的不同吸附剂于1.5ml子弹头离心管,加入0.01mg/l的混合标准溶液1ml,涡旋振荡2min,12000r/min离心2min后,取上清液过0.22μm有机滤膜过滤后上机检测。每种吸附剂做三个平行,计算平均回收率,实验结果表明,psa,c
18
,无水mgso4对砜吡草唑的吸附回收率都在95~105%之间;说明三种物质均不会对砜吡草唑造成吸附。
[0052]
2.2.3吸附剂组合用量的确定
[0053]
植物源性样品使用quechers法进行提取和净化时通常会使用两种或多种吸附净化剂,以较好的除去样品中的有机酸、脂类、糖类等干扰仪器分析的杂质。实验复配了三组吸附剂(i:10mgpsa 10mgc
18
50mg无水mgso4,ii:20mgpsa 20mgc
18
50mg无水mgso4,iii:50mgpsa 50mgc
18
50mg无水mgso4)对小麦空白样品进行0.02mg/kg加标回收实验,考察不同吸附剂组合净化效果,实验结果如图4所示。实验结果表明复配的三组吸附剂组合都可以满足实验回收率的要求,其回收率范围都在90~110%之间;从实际净化效果看,经ii,iii型吸附剂组合的除杂净化后,净化效果较好,对目标农药影响较小。综合考虑净化效果、成本、便捷、仪器分析时干扰杂质及基线,选择20mgpsa 20mgc
18
50mg无水mgso4确定为最终实验的复配吸附净化剂的种类和用量。
[0054]
2.2.4基质效应的影响
[0055]
基质效应(matrix effects,me)是指待测物质的样品共提取物,会增强或抑制待测物的检测响应,从而对检测结果的准确度和精密度所造成影响。基质效应在气相系统中主要表现为“基质诱导色谱响应增强现象”,即基质增强效应。基质效应影响大时会降低方法的灵敏度和影响方法的准确性,给测定带来误差,因此,方法开发和确证过程中需要对基质效应(matrix effects,me)做出评价。基质效应与基质的种类及基质干扰物的含量、分析物的特性、色谱分离条件、不同仪器离子源设计等有关,可按以下公式对基质效应进行量化评估:基质效应(me,%)=[(基质匹配校准曲线斜率/纯溶剂标准曲线斜率)-1]
×
100。|me|<20%为弱基质效应,可忽略而无需采取补偿措施;20%≤|me|≤50%为中等程度基质效应,|me|>50%为强基质效应,须采取措施补偿基质效应。试验按照1.2.2的前处理方法制备空白基质溶液,按照1.2.1的方法配制标准工作溶液和基质匹配标准工作溶液。按照以上公式计算不同基质中的基质效应如图5所示。
[0056]
由图5可以看出,砜吡草唑在8种基质中基质效应均为中等附近,应考虑采用措施补偿基质效应。在农药残留分析检测过程中,采用基质匹配标准溶液进行定量分析可以克服基质效应对定量结果的影响。我国关于植物源性食品农药残留检测的国家标准gb/t 20769-2008及gb 23200.113-2018等都规定了基质匹配标准溶液的定量方法,欧盟在食品中农药残留分析方法的指导性文件sante/12682/2019中也推荐使用基质匹配标准溶液进行定量分析。然而,针对多种多样的样品基质,在配制基质匹配标准溶液的过程中,大大降低了检测效率。通过图5可知不同类型样品中基质效应差别较小,所以在实际操作中,可选择小麦为代表基质配制基质匹配标准工作溶液,克服基质效应,在保证定量准确性和稳定性的前提下,提高检测效率。
[0057]
2.3方法学验证
[0058]
2.3.1线性范围、相关系数和方法检测限
[0059]
用空白基质溶液配制浓度为0.5μg/l、2μg/l、5μg/l、10μg/l、20μg/l和25μg/l系列砜吡草唑基质匹配标准工作溶液,按1.2.3进样检测,以砜吡草唑浓度(x,ng/ml)为横坐标,以砜吡草唑峰面积为纵坐标绘制基质标准工作曲线,得到线性方程和相关系数;在空白样品溶液中添加适量的标准溶液后上机测定,以s/n=10确定定量限(loq),相关数据见表1。
[0060]
2.3.2回收率
[0061]
分别对稻谷、小麦、高粱、玉米、大豆、花生、甘薯、马铃薯和葵花籽空白样品进行了不同浓度的标准添加回收实验,标准添加回收实验覆盖定量限,主要mrl值,每个加标水平测6次平行,砜吡草唑在这些植物源样品中的添加浓度及回收率的实验数据见表2。
[0062]
表2砜吡草唑在不同样品中的加标回收率和相对标准偏差(n=6)
[0063][0064][0065]
2.4实际样品检测
[0066]
用本方法对50个稻谷、小麦、高粱、玉米、大豆、花生、甘薯、马铃薯和葵花籽进行检测,检测结果均为未检出。
[0067]
3结论
[0068]
首次建立了气相色谱三重四极杆质谱法测定植物源食品中砜吡草唑残留量的检测方法,在优化实验条件下,目标化合物在0.001-0.05mg/kg范围内的线性关系良好,相关系数大于0.99。空白样品在低、中、高4个添加水平下的平均回收率为83.8%-104.3%,相对标准偏差(n=6)为2.6%-6.7%,方法定量限为0.001mg/kg,方法灵敏度高、操作简单、快速、准确、成本低,能满足植物源性食品中残留的砜吡草唑检测需求。
[0069]
以上对本发明的实施例进行了详细说明,但所述内容仅为本发明的较佳实施例,并不用以限制本发明。凡在本发明的申请范围内所做的任何修改、等同替换和改进等,均应包含在本发明的保护范围之内。
再多了解一些
本文用于企业家、创业者技术爱好者查询,结果仅供参考。